










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIngeniería y competitividad
Print version ISSN 0123-3033
Ing. compet. vol.17 no.1 Cali Jan./June 2015
Remote homology detection of proteins using 3D models enriched with physicochemical properties
Detección de homología remota de proteínas usando modelos 3D enriquecidos con propiedades fisicoquímicas
Oscar F. Bedoya*
E-mail: oscar.bedoya@correounivalle.edu.co
Irene Tischer*
E-mail: irene.tischer@correounivalle.edu.co
* Escuela de Ingeniería de Sistemas y Computación, Universidad del Valle, Cali, Colombia.
Eje temático: SYSTEMS ENGINEERING/INGENIERIA DE SISTEMAS
Recibido: Mayo 26 de 2014
Aceptado: Enero 29 de 2015
Abstract
In this paper, a new method for remote protein homology detection called remote-3DP, is presented. The remote-3DP method is based on both predicted 3D information and physiochemical properties of amino acids. The remote-3DP method considers only 10 structural models to represent a protein and to distinguish between remote homologues and non-remote homologues in 54 SCOP families. The low dimensionality of the protein representation allows us to use different classification techniques and discover which one works better for each SCOP family. In this paper, we show that including a physicochemical property along with predicted 3D information into a local structural element, actually improves the accuracy in remote homology detection. The highest ROC score for a set of models that includes 3D information and the Hydropathy index reaches 0.953 on the SCOP 1.53 dataset. In addition, a model that ensembles the outputs of 10 physicochemical properties is built to make a consensus decision. The consensus strategy reaches a ROC score of 0.963 on the SCOP 1.53 dataset, surpassing the current methods based on sequence composition which accuracy range from 0.87 to 0.92.
Keywords: Classification, physicochemical properties, remote homology detection, SCOP family, 3D structural models.
Resumen
En este artículo se presenta un nuevo método para la detección de homología remota llamado remote-3DP. El método remote-3DP se basa tanto en información 3D predicha como en las propiedades fisicoquímicas de los aminoácidos. El método considera tan sólo 10 modelos estructurales para representar una proteína y distinguir los homólogos remotos de los no remotos en 54 familias SCOP. La baja dimensionalidad de la representación permite usar diferentes técnicas de clasificación y descubrir cuál funciona mejor para cada familia. En este artículo, se muestra que al incluir una propiedad fisicoquímica junto con la información 3D en un elemento estructural local, de hecho mejora la exactitud de la detección de homología remota. El puntaje ROC para un conjunto de modelos que incluye el índice de hidropatía alcanza un puntaje de 0. 953 para el conjunto de datos SCOP 1.53. Además, se propone un modelo de ensamble que utiliza las salidas obtenidas para las 10 propiedades y así tomar una decisión consenso. La estrategia consenso alcanza un puntaje ROC de 0.963 sobre el conjunto de datos SCOP 1.53, sobrepasando los métodos actuales basados en la composición de la secuencia cuya exactitud varía de 0.87 a 0.92.
Palabras clave: Clasificación, detección de homología remota, familia SCOP, modelos estructurales 3D propiedades fisicoquímicas.
1. Introduction
Remote homology detection problem is about identifying proteins that are functionally and structurally related but at the same time do not share sequence similarity. The problem can be defined using the SCOP hierarchy. Considering the four levels of the SCOP hierarchy (i.e., class, fold, superfamily and family), the remote homologs of a protein P in family F are proteins in the same superfamily of P that do not belong to F. According to the definition of a SCOP superfamily, the remote homologs of P are proteins that have a common ancestor and thus they still share function and structure. However, remote homologs of P belong to a family different from F, which implies that P and its remote homologs do not share sequence similarity. According to Vendruscolo & Dobson (2005) and Muda et al. (2011), finding remote homologs for a target protein P is considered a fundamental step in biomedical applications such as drug discovery, where proteins that share common functions given a specific protein sequence have to be identified.
Several methods have been proposed to determine remote homology, such as SVM-I-sites by Hou et al. (2003), SVM-RQA by Yang et al. (2008), SVM-PCD by Webb-Robertson et al. (2010), BioSVM-2L by Muda et al. (2011), and SVM-PDT by Liu et al. (2012). Some of the most recent works are sequence composition-based methods, which are based on using subsequences, motifs or word similarity from protein sequences to extract features that help discriminating protein families. The ROC score (Receiver Operating Characteristic) of these methods ranges from 0.87 to 0.92. There are sequence composition-based methods that incorporate physicochemical properties of amino acids. The SVM-PDT method proposed by Liu et al. (2012) considers the distance between the physicochemical values of two amino acids separated by residues along the protein chain. Liu et al. (2012) uses eight values, which means eight separation values starting with the distance between a residue ri and ri+1 until ri and ri+8. The eight values are calculated using a single physicochemical property. SVM-PDT considers 531 physicochemical properties. Thus, a total of 531·8=4248 values are calculated in the vector representation.
In this paper, we propose a new method that uses models based on both predicted 3D information and physicochemical properties of amino acids. Different sets of models are presented, each collection of models uses a specific physicochemical property. In addition, a model that ensembles the outputs of the individual collections is built to make a consensus decision. In the following section every step in the remote-3DP method is explained in detail. In Section 3, the results are given considering the SCOP 1.53 benchmark. Finally, the conclusions of the research are presented in Section 4.
2. Methodology
The remote-3DP method (3D enriched with physicochemical properties) is divided in three steps. First, 3D models that incorporate physicochemical properties are obtained; different collections of models are presented. Then, the models are used to represent every protein in the dataset and a classifier is built for each SCOP family. Finally, a consensus model is built to improve the accuracy of remote homology detection.
2.1 Obtaining the models
The first step in the remote-3DP method is obtaining models from the 3D predicted information and the physicochemical properties of amino acids. Given an amino acid sequence, the 3D information (i.e., contact map) is predicted using the NNcon 1.0 program proposed by Cheng et al. (2009). The NNcon1.0 uses a neural network to predict the general contact map and another neural network to predict the beta-sheet contacts. The NNcon1.0 is available at http://sysbio.rnet.missouri.edu/multicom_toolbox/tools.html. A contact map is a matrix obtained by binarily discretizing each value in the distance matrix (i.e., 1: contact, 0: non-contact). A distance matrix of a protein is a square matrix containing the Euclidean distances between all pairs of Cα atoms in the protein. In this research, we assume that two residues are in contact if the Euclidean distance between the corresponding Cα atoms is less o equals to 8.0 Angstroms.
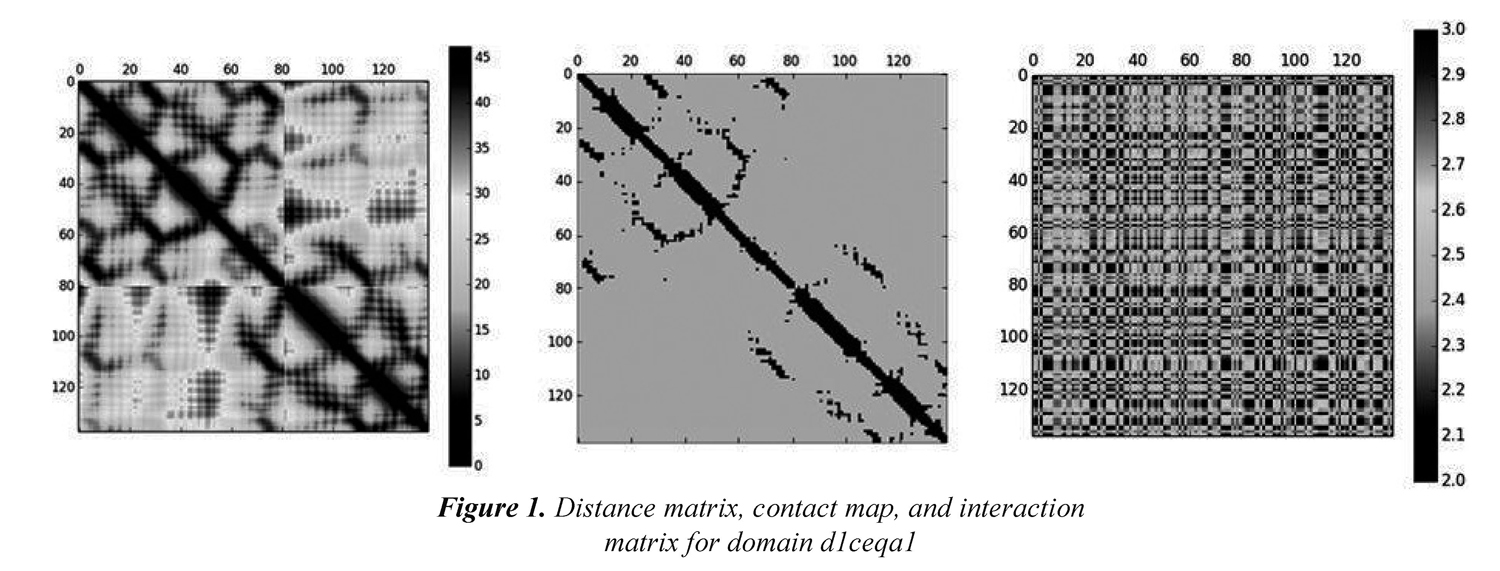
We also included physicochemical properties in the process of obtaining the models that will be used in remote homology detection. Every amino acid index contains 20 values representing a particular physical o biochemical property of amino acids. According to Yang et al. (2008), the hypothesis behind incorporating physicochemical properties into the definition of models in remote homology detection is that because protein structure and function are conserved during evolution, the similarity between two distantly related proteins may lie in the physicochemical properties of the amino acids rather than the sequence identities. In this paper, the physicochemical properties are used to represent the interactions between every pair of amino acids in a protein. We define an interaction matrix Ip as a square matrix that holds the additions between the values of the physicochemical property p for each pair of amino acids. First, the 20 values of each physicochemical property have to be scaled to a range of values that make them comparable to the values in the contact maps. We chose to scale every value of each physicochemical property to the range [1,1.5] considering the values in the contact map (i.e., 1 and 0). Then, every position (i,j) in the matrix IP is calculated as the addition between the values of the physicochemical property p for residues at positions i and j. Figure 1(a) shows the distance matrix for domain d1ceqa1; the bar scale shows the distance in Angstroms. Figure 1(b) shows the contact map for the same domain; the contacts (i.e., Cα atoms whose distance is less o equals to 8.0 Angstroms) are shown in black and non-contacts are shown in gray, and Figure 1(c) shows the corresponding interaction matrix Ip when the Hydrophobicity index is considered.
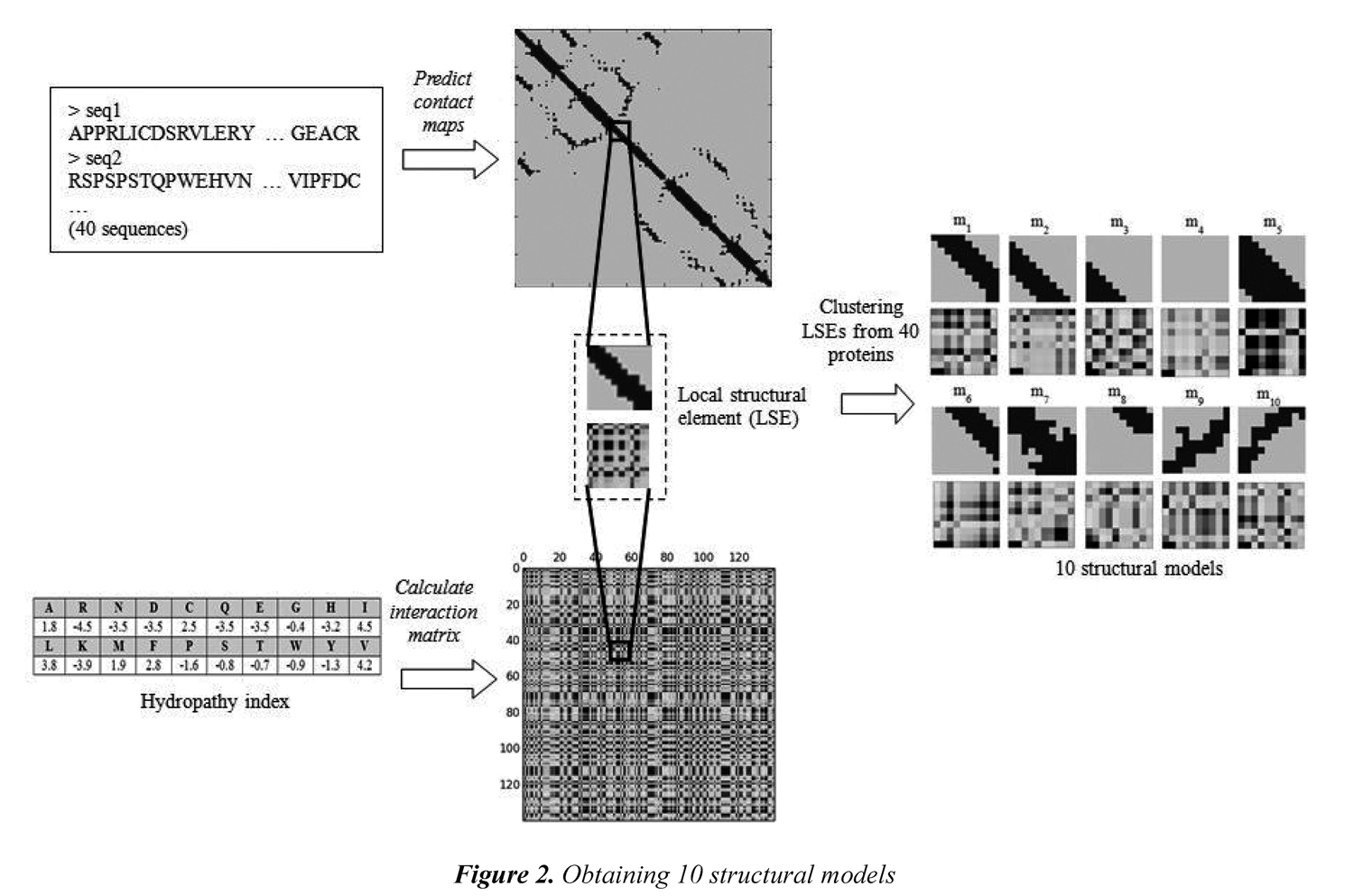
A collection of structural models that incorporates 3D information (i.e., common 10x10 submatrices in the contact maps) along with physicochemical properties (i.e., the corresponding 10x10 submatrices in the interaction matrix) are used in the remote-3DP method. The models are extracted from a training dataset of 40 proteins selected from the SCOP 1.55. The proteins were selected by taking two proteins for each of the 20 SCOP superfamilies in the dataset. Obtaining the structural models starts by calculating a contact map for each protein in the training dataset (i.e., by using the NNcon1.0 program). Then, submatrices of 10x10 are extracted from the contact map as local structural features. The size of the submatrix was taken just as Choi et al. (2004) who tried submatrices from 8x8 up to 16x16 and found that size 10x10 allow to capture 3D interactions that occur in the distance matrix. In addition, 10x10 submatrices are also extracted from the interaction matrix to create a local structural element (LSE) formed by 200 values. Each local structural element includes both 3D structure and values from a given physicochemical property. After having a collection of local structural elements, the clustering algorithm CLARA (Clustering Large Applications) was used to obtain the representative LSEs (i.e., resulting medoids after clustering algorithm) that are taken as structural models in the remote-3DP method.
The CLARA algorithm is a clustering strategy that starts by taking a sample from the dataset and then uses the PAM algorithm (Partitioning around medoids). Using a sampling strategy allows the CLARA algorithm handling large datasets. In this research, we used the implementation of the CLARA algorithm available in the R language (http://www.r-project.org/). It is expected that the local structural elements taken as models after the clustering algorithm are commonly and frequently used in several proteins. Following the methodology proposed by Choi et al. (2004), each protein in training is first clustered into 50 representative LSEs, and then the obtained medoids are clustered again to obtain a final set of k models. This allows having a reasonable amount of submatrices in the clustering process. In this paper, we used k=10 to obtain the final number of models. Figure 2 shows the process of obtaining 10 structural models from the amino acid sequences.
Each interaction matrix uses only one physicochemical property. In this paper, we propose to use 10 different physicochemical properties to analyze their effect in the process of obtaining the models. We selected the 10 physicochemical properties related in the state of the art that have shown the strongest relationship with the three-dimensional structure of proteins. According to Grigoriev & Kim (1999), the three-dimensional structure of a protein is determined by the physicochemical properties of its residues and we pretend to discover if including the physicochemical properties in the model definition has an impact in remote homology detection. The 10 selected physicochemical properties are: Hydropathy index, Polarity, Normalized van der Waals volume, Atom-based hydrophobic moment, The Kerr-constant increments, Spin-spin coupling constants 3JHalpha-NH, The Chou-Fasman parameter of the coil conformation, Alpha-helix propensity derived from designed sequences, Relative preference value at C', and pK (-COOH)
Figure 3 shows the 10 models that are obtained when the physicochemical property Hydropathy is used in the interaction matrix; we call this set of models the Hydropathy collection. Every model reflects local structural interactions. m1, m2, and m6 represent different positions of a beta sheet. m3 is the outer part (right) of a helix. m4 represents the non-contacts between residues and it is the most frequent model in any protein. m5 is common model in the diagonal of a helix. m7 is a model that can occur in both helix and beta sheet. m8 is the outer part (left) of a helix. m9 and m10 represent the anti-parallel beta-sheets. A total of 10 different collections of structural models were obtained. Each collection uses one of the 10 selected physicochemical properties.

2.2 Building a classifier for each SCOP family
We define a count vector of a protein as the set of values indicating the times that each structural model is identified in the local structural elements of a protein. The size of the count vector is 10 because we decided to keep that number of models and vary the physicochemical properties. The count vector is obtained from the predicted contact map and the interaction matrix; both of them are calculated from the primary sequence.
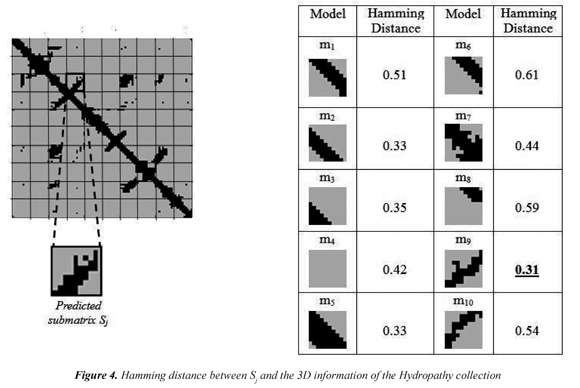
Given a predicted contact map (PCM) of size nxn, overlapping submatrices of 10x10 are extracted. At the same time overlapping submatrices of 10x10 are extracted from the interaction matrix Ip. Each local structural element has 200 values, 100 values from the 10x10 submatrix of the PCM and 100 values from the 10x10 submatrix of the interaction matrix. Each local structural element Sj in a protein is assigned to the closest model mi in a given collection by using a combination of the normalized Hamming distance and the Euclidean distance. First, the normalized Hamming distance is used to compare the submatrices from the predicted contact map to the 3D information in the models. The Hamming distance is used because each submatrix in the contact map is a set of discretized values that describes a particular shape of 3D local structural interactions and thus the Hamming distance captures the model's similarity. Figure 4 shows the Hamming distance between a submatrix Sj and the 3D information of the models in the collection. Then, the Euclidean distance is used to compare the submatrices from the interaction matrix to the physicochemical properties values that are part of the models. A different metric is used because submatrices from the interaction matrix are composed by real numbers instead of the discretized values in the predicted contact map. As might be expected, different decisions can be taken when the closest model is calculated for a structural element Sj using the contact map and the interaction matrix information even for the same local structural element. Thus, the final decision about the closest model of a local structural element Sj is taken by normalizing the Hamming and Euclidean distances, and then adding the two values.
After calculating the count vector for each protein in the dataset, a normalized count vector (NCV) is obtained. The normalization assures that every value in the count vector contributes equally. The normalized count vector of the k-th protein in a dataset is represented as the vector NCVk=[Ak1,Ak2,…,AkM], where Akm is the normalized count value for the m-th model in the k-th protein and is defined in Eq. (1).
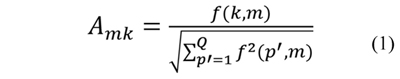
where f(k,m) is the m-th value in the count vector of the k-th protein and Q is the number of proteins in the dataset. The normalized count vector of the k-th protein is the vector NCVk and contains the normalized counts of the overlapping predicted submatrices.
Once the normalized count vector is calculated for each protein in the dataset, we propose to use different classification techniques to distinguish remote homologs from non-remote homologs in a given SCOP family. In remote homology detection a classifier is trained for each SCOP family. For each family, the proteins within the family are taken as the positive testing set while proteins outside the family but within the same superfamily are considered as the positive training set. Negative examples are taken from proteins outside the family's fold. For instance, when a classifier is built for SCOP family b.1.1.1, proteins in families b.1.1.2, b.1.1.3, b.1.1.4, and b.1.1.5 are taken as the positive training dataset because they belong to the superfamily b.1.1. Proteins in family b.1.1.1 are used as the positive test set. Finally, proteins outside the fold b.1 are taken as negative examples. We propose to use the classifiers in the WEKA data mining software developed by Hall et al. (2009) to identify remote homologs for each family. The WEKA program (http://www.cs.waikato.ac.nz/ml/weka/) has several classification techniques that can be applied when a classifier is built for each SCOP family. There are at least 30 classification algorithms including different strategies such as Bayes, Functions, Miscellaneous, and Decision trees methods. In this paper, remote homology detection is performed by selecting a classifier for each SCOP family using 5-fold cross validation on the training dataset. In the 5-fold cross validation strategy the training set is divided in five parts. One part is used as the validation data and the remaining k-1 parts are used as training data. The process is repeated five times.
2.3 Consensus strategy in remote homology detection
A total of 10 collections of models were considered. Each collection uses a different physicochemical property. In this paper, a consensus model is built to make a better decision that ensemble the outputs of the 10 collections. The classifier built for each SCOP family produces not only a class label indicating whether a target protein is remote to the family or not, but also a score. The score is a numerical value produced by the classifier and it ranges from 0.0 to 1.0. The closer to 1.0 the score, the bigger the probability of getting a +1 class label (i.e., remote to the family), and the closer to 0.0 the score, the bigger the probability of getting a -1 class label (i.e., non-remote to the family). We propose to build a model that takes the scores from the 10 classifiers (i.e., one from each collection) and produces a consensus decision. Obtaining a consensus decision for a given protein P during testing starts by calculating 10 scores from the 10 classifiers (i.e., each classifier is based on a different physicochemical property). Then, the scores are given to the consensus model, which produces a final decision. An important step when a consensus strategy is built is about training the consensus model. The consensus model was trained with the scores of the 10 classifiers using 5-cross validation technique on the training dataset. In that way, the testing dataset was kept unseen during the training of the consensus model.
3. Results and discussion
In this section, the results of the experiments are shown. A comparison with some of the current methods is shown.
3.1 Measuring the accuracy
In this paper, the ROC score (receiver operating characteristic) is used to measure the accuracy of the remote-3DP method. The ROC score is the normalized area under the curve, which exhibits the relationship between true positives and false positives for different classification thresholds.
3.2 Selecting the dataset
In this paper the ASTRAL SCOP database 1.53 is used for testing. The SCOP 1.53 has been used as a standard for remote homology detection. A total of 54 families and 4352 sequences are considered.
3.3. Evaluating the remote-3DP method
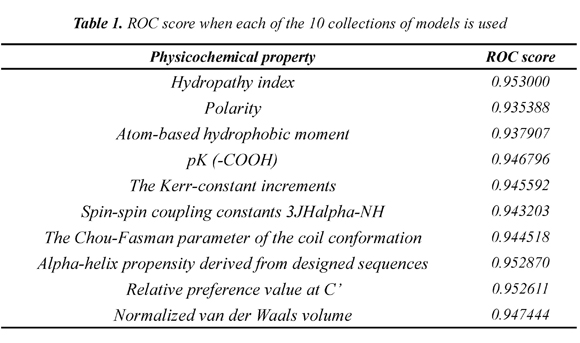
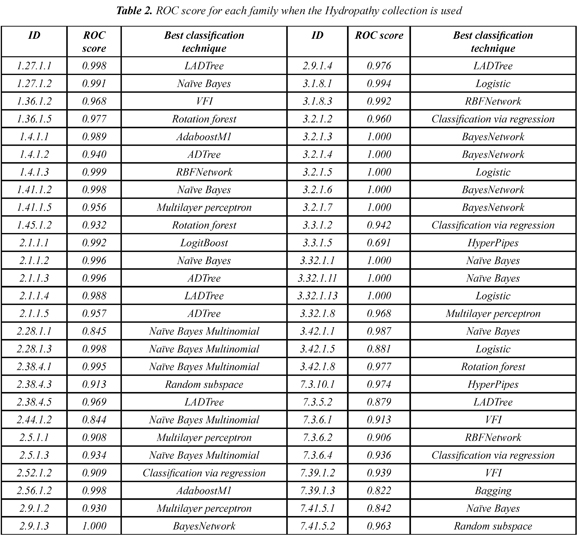
Table 1 shows the mean ROC score over the 54 families when each of the 10 collections of models is used. The Hydropathy collection is the most accurate collection with a ROC score of 0.953. In addition, we found that each SCOP family has a classifier that separates better remote homologs from non-remote homologs. Table 2 shows the best classifier for each of the 54 SCOP families when the Hydropathy collection is used.
3.3.1.Evaluating the effect of incorporating physicochemical properties
A key aspect about incorporating physiochemical properties in the definition of models was to discover if it helps to obtain models that discriminate remote homologues better. We compare the collections of models that use 3D information and physicochemical properties with models that only use 3D information. Obtaining models based on 3D information is done by using only the discretized submatrices of 10x10 from the contact maps. The mean ROC score when the 3D collection is used reaches 0.941574074, which only surpasses the Polarity and Atom-based hydrophobic moment collections with 0.935388 and 0.937907, respectively. Eight out of the 10 collections used in this paper reach a higher mean ROC score than the 3D collection. The results show that including physiochemical properties can actually improve the quality of the models in remote homology detection.
3.3.2 Evaluating the consensus model
The consensus model was built using three classifiers: Bayesnet, NaiveBayes, and VFI (Classification by voting feature intervals). We found that each SCOP family has a classification technique that should be used as its consensus model. Using the consensus model, a mean ROC score of 0.962926 is obtained. The ROC score of the consensus model surpasses even the highest individual collection of models. There are collections of models that work better for some families and according to the results the consensus model is actually able to recognize that pattern and produce a better classification decision for each family.
3.3.3 Comparison with recent discriminative methods
We compared the remote-3DP method with some of the most recent discriminative strategies, such as SVM-RQA by Yang et al. (2008), SVM-PCD by Webb-Robertson et al. (2010), SVM-PDT by Liu et al. (2012), SVM-WCM by Lingner & Meinicke (2008), and SVM-LA by Saigo et al. (2004), which ROC scores are 0.912, 0.906, 0.916, 0.904, 0.925, respectively. The remote-3DP was tested on the SCOP 1.53 dataset, which is the same dataset used in the current methods. When the remote-3DP method uses the Hydropathy collection, it reaches the highest accuracy for a remote homology detection method based on the sequence composition (i.e., 0.953). The main differences between the remote-3DP method and the existing strategies are: the low dimensionality of the vector representation (i.e., only 10 values), incorporating 3D and physicochemical properties in the structural elements, and using a consensus strategy.
4. Conclusions
In this paper, we proposed a new method that uses models based on both 3D information and physicochemical properties of amino acids. The remote-3DP method considers only 10 structural models to represent a protein and distinguish between remote homologues and non-remote homologues in 54 SCOP families. The low dimensionality of the protein representation allows us to use different classification techniques and discover which one works better for each SCOP family. We found that including a physicochemical property along with 3D information in a local structural element, actually improves the accuracy in remote homology detection.
In addition, a model that ensembles the outputs of the 10 collections is built to make a consensus decision. The numerical score of each collection is used to feed a consensus model that is able to identify which physicochemical property works better for the proteins into a SCOP family. The consensus model reaches a ROC score of 0.963. Both the remote-3DP with the Hydropathy collection and remote-3DP with the consensus model surpass the current methods in remote homology detection based on sequence composition. Different physicochemical properties might be tested on the remote-3DP method. We selected just some of the physicochemical properties that have shown a high relationship to the 3D structure of the protein. Considering that there are 544 physicochemical properties in the AAindex, several more indices could be tested. It is expected that some physicochemical properties work better for some families, and thus, the consensus strategy that takes the outcomes of more collections of models might reach an even higher accuracy.
5. References
Cheng, J., Tegge, A., Wang, Z., & Eickholt, J. (2009). NNcon: improved Protein Contact Map Prediction Using 2D-Recursive Neural Networks. Nucleic Acids Research, 37 (1), 515-518. [ Links ]
Choi, I.-G., Kwon, J., & Kim, S. (2004). Local Feature Frequency Profile: A Method to Measure Structural Similarity in Proteins. Proceedings of the National Academy of Sciences of the United States of America, 101 (11), 3797-3802. [ Links ]
Grigoriev, I. & Kim, S. (1999). Detection of protein fold similarity based on correlation of amino acid properties. Proceedings of the National Academy of Sciences of the United States of America, 96 (25), 14318-14323. [ Links ]
Hall, M., Frank, E., Holmes, G., Pfahringer, B., Reutemann, P., & Witten, I. (2009). The WEKA Data Mining Software: an update. ACM SIGKDD Explorations, 11 (1), 10-18. [ Links ]
Hou, Y., Hsu, W., Lee, M., & Bystroff, C. (2003). Efficient Remote Homology Detection Using Local Structure. Bioinformatics, 19 (17), 2294-2301. [ Links ]
Lingner, T., & Meinicke, P. (2008). Word correlation matrices for protein sequence analysis and remote homology detection. BMC Bioinformatics, 9 (1), 1730-1743. [ Links ]
Liu, B., Wang, X., Chen, Q., Dong, Q., & Lan, X. (2012). Using Amino Acid Physicochemical Distance Transformation for Fast Protein Remote Homology Detection. PLoS ONE, 7 (9), 1-10. [ Links ]
Muda, H., Saad, P., & Othman, R. (2011). Remote protein homology detection and fold recognition using two-layer support vector machine classifiers. Computers in Biology and Medicine, 41 (1), 687-699. [ Links ]
Saigo, H., Vert, J., Ueda, N., & Akutsu T. (2004). Protein Homology Detection Using String Alignment Kernels. Bioinformatics, 20 (1), 1682-1689. [ Links ]
Vendruscolo, M., & Dobson, C. (2005). Towards complete descriptions of free energy landscapes of proteins. Philosophical transactions of the royal society, 363 (1), 433-452. [ Links ]
Webb-Robertson, B., Ratuiste, K., & Oehmen, C. (2010). Physicochemical property distributions for accurate and rapid pairwise protein homology detection. BMC Bioinformatics, 11 (1), 145-183. [ Links ]
Yang, Y., Tantoso, E., & Li, K. (2008). Remote protein homology detection using recurrence quantification analysis and amino acid physicochemical properties. Journal of Theoretical Biology, 252 (1), 145-154. [ Links ]

Revista Ingeniería y Competitividad por Universidad del Valle se encuentra bajo una licencia Creative Commons Reconocimiento - Debe reconocer adecuadamente la autoría, proporcionar un enlace a la licencia e indicar si se han realizado cambios. Puede hacerlo de cualquier manera razonable, pero no de una manera que sugiera que tiene el apoyo del licenciador o lo recibe por el uso que hace.
