










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.27 supl.1 Bogotá Dec. 2020 Epub Aug 24, 2021
https://doi.org/10.1016/j.rcreu.2020.05.026
Artículo de revisión
Tratamiento farmacológico de la hipertensión pulmonar asociada a la esclerosis sistémica: revisión sistemática de la literatura
aSection of Rheumatology, Department of Internal Medicine, Hospital Universitario Nacional, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
bREUMAVANCE Group, Section of Rheumatology, Department of Internal Medicine, Fundación Santa Fe de Bogotá University Hospital, Bogotá, D.C., Colombia
cSection of Cardiology, Department of Internal Medicine, Hospital Universitario Nacional, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
dFernando Chalem Institute of Rheumatology, Bogotá, D.C., Colombia
Antecedentes:
La esclerosis sistémica es una enfermedad autoinmune que puede afectar significativamente los órganos internos, aumentando la mortalidad asociada con dicho trastorno. La hipertensión pulmonar es una de las afecciones que aumentan la morbimortalidad de la enfermedad. El objetivo del estudio fue desarrollar una revisión sistemática de la literatura sobre los diferentes tratamientos farmacológicos para la hipertensión pulmonar asociada a la esclerosis sistémica.
Materiales y métodos:
Se hizo una revisión sistemática de la literatura en Cochrane, PubMed y EMBASE. Se incluyeron ensayos clínicos aleatorizados controlados, ensayos clínicos controlados, así como estudios de cohortes y de casos y controles utilizando alguno de los tratamientos disponibles, tales como los análogos de la prostaciclina, los antagonistas de los receptores de endotelina, los inhibidores de la fosfodiesterasa o los estimulantes de la guanilato ciclasa soluble. Se evaluó la calidad metodológica de los estudios utilizando la metodología Cochrane para los ensayos clínicos, y se aplicó la herramienta SIGN para los estudios de cohortes y de casos y controles.
Resultados:
Inicialmente se encontraron 870 estudios, de los cuales finalmente se incluyeron 20 en la revisión sistemática, después de aplicar los criterios de inclusión y exclusión. La mayoría de los estudios incluyeron el bosentán en las estrategias de tratamiento (n = 13). Además, la mayoría de los estudios tomaron datos de los ensayos clínicos más importantes sobre el tratamiento farmacológico de la hipertensión pulmonar de diversas etiologías. Algunos estudios que utilizaron el bosentán no suministraron información sobre datos relacionados con algunos resultados. La mitad de los estudios seleccionados fueron ensayos clínicos, de los cuales dos fueron aleatorizados. La calidad de estos estudios fue buena. La otra mitad incluyó estudios observacionales, con evidencia de calidad moderada. En general, se encontró que los medicamentos aprobados mejoran la distancia en la caminata de seis minutos, la clase funcional de la OMS, la clasificación funcional de la NYHA, o el puntaje de disnea en la escala de Borg, dependiendo de la dosis administrada en los diferentes estudios, y también los desenlaces compuestos que incluyen hospitalizaciones, empeoramiento clínico, muerte, etc. En cuanto a la mortalidad, se encontraron diferencias significativas en las tasas de supervivencia al comparar los medicamentos aprobados versus placebo, siendo las terapias combinadas superiores a la monoterapia. Las mediciones de variables hemodinámicas tales como el índice cardíaco, las resistencias pulmonares y las presiones pulmonares mejoraron. Además, los niveles de NT-pro BNP se redujeron, con algunas excepciones.
Conclusiones:
En la mayoría de los estudios con buena calidad de evidencia, se encontró una mejoría en los desenlaces clínicos y no clínicos, que favorece el uso de los medicamentos aprobados para el tratamiento de la hipertensión pulmonar asociada a la esclerosis sistémica.
Palabras clave: Esclerosis sistémica Escleroderma; Hipertensión pulmonar; Tratamiento
Background:
Systemic sclerosis is an autoimmune disease that could significantly affect internal organs, increasing the mortality associated with the disorder. Pulmonary hypertension is one of the conditions that increase the morbidity and mortality of the disease. The aim of the study was to develop a systematic literature review about the different pharmacological treatments for systemic sclerosis-associated pulmonary hypertension.
Materials and methods:
A systematic literature review was made on Cochrane, PubMed and EMBASE. Randomized controlled clinical trials, controlled clinical trials, cohort and case-control studies using any of the available treatments such as prostacyclin analogs, endothelin receptor antagonists, phosphodiesterase inhibitors or soluble guanylate cyclase stimulants were included. The methodological quality of the studies was assessed using Cochrane methodology for clinical trials, and the SIGN tool was applied for cohort and case-control studies.
Results:
Initially, 870 studies were found, and 20 studies were finally included in the systematic review after applying inclusion and exclusion criteria. Most studies included Bosentan in the treatment strategies (N = 13). Also, most studies took data from the most important clinical trials for pharmacological treatment of pulmonary hypertension of several etiologies. Some studies using Bosentan did not give information about data regarding some outcomes. Half of the selected studies were clinical trials, of which two were randomized. The quality of these studies was good. The other half were observational studies, with moderate quality of evidence. In general, it was found that approved medications improve the 6-minute walk distance, WHO functional class, NYHA functional classification, or Borg dyspnea score, depending on the given dose in several studies, and also compound outcomes that include hospitalizations, clinical worsening, death, etc. Regarding mortality, there were significant differences in survival rates when comparing approved medications vs. placebo, being combined therapies superior to monotherapy. Measurements of hemodynamic variables such as cardiac index, pulmonary resistances, and pulmonary pressures were improved. Also, NT-proBNP levels were reduced, with some exceptions.
Conclusions:
In most studies with good quality of evidence, it was found an improvement in clinical and non-clinical outcomes, favoring the use of approved medications for the treatment of systemic sclerosis-associated pulmonary hypertension.
Keywords: Systemic sclerosis; Scleroderma; Pulmonary hypertension; Treatment
Introducción
La esclerosis sistémica (SSc) es una enfermedad autoinmune asociada al tejido conectivo, que se caracteriza por el desarrollo de lesión microvascular, fibrosis generalizada con compromiso multiorgánico, y una regulación inadecuada del sistema inmunológico. La prevalencia mundial de la enfermedad es variable dependiendo de la población estudiada. La enfermedad es menos frecuente en poblaciones del norte de Europa y Japón, mientras que en el sur de Europa, Norteamérica y Australia es más prevalente. Del mismo modo que otras enfermedades autoinmunes asociadas al tejido conectivo, es más frecuente en las mujeres. Sin embargo, la enfermedad tiene un peor pronóstico en los hombres, ya que está asociada con un mayor riesgo de compromiso cutáneo difuso y de mortalidad1.
Según la extensión del compromiso cutáneo, la enfermedad se clasifica en: SSc limitada, SSc difusa y SSc sin esclerodermia (sine). Adicionalmente, puede presentarse como un síndrome de superposición, en el que están presentes algunas características clínicas de otras enfermedades auto-inmunes asociadas con el tejido conectivo, tales como el lupus eritematoso sistémico. Considerando la historia natural de la enfermedad, se ha determinado que la extensión del compromiso cutáneo y la tasa de progresión son marcadores de un posible compromiso de los órganos internos, incluyendo el esófago, los pulmones, los riñones, etc.1.
El compromiso pulmonar es una de las complicaciones más frecuentes, y tiene un alto impacto en la morbimortalidad. Las principales complicaciones pulmonares que se pueden desarrollar en estos pacientes son la fibrosis (asociada con la tendencia de la enfermedad a generar un proceso aberrante de cicatrización de las heridas) y la hipertensión pulmonar (asociada con vasculopatía proliferativa)2. Las complicaciones pulmonares han reemplazado a la crisis renal como la principal causa de mortalidad en los pacientes con SSc.
Hipertensión pulmonar asociada a la esclerosis sistémica
La hipertensión pulmonar asociada a la esclerosis sistémica (SSc-HTP) es una complicación frecuente y severa en estos pacientes. Afecta aproximadamente al 10% de los pacientes, y se asocia con una baja supervivencia desde el diagnóstico, que es de aproximadamente tres anos3.
La hipertensión pulmonar se definió como una presión media en la arteria pulmonar por encima de 25 mmHg, y una presión en cuna de la arteria pulmonar inferior a 15 mmHg, medidas mediante cateterismo cardíaco derecho, en ausencia de enfermedad pulmonar intersticial4.
La fisiopatología de la enfermedad es muy compleja, y abarca principalmente el desarrollo de lesiones vasculares secundarias a lesión del endotelio pulmonar y disfunción endotelial, produciendo una fibrosis aberrante y una reparación vascular inadecuada, que da lugar a una vasculopatía obliterante. Las arteriolas presentan un engrosamiento importante debido a un aumento del tejido muscular y cambios inflamatorios perivasculares asociados a una función inadecuada de los monocitos/macrófagos. Adicionalmente, la presencia de disfunción endotelial y estrés oxidativo contribuye en gran medida a la patogénesis de la enfermedad2.
La Organización Mundial de la Salud clasifica la hipertensión pulmonar en cinco categorías según las características clínicas y fisiopatológicas:
Hipertensión arterial pulmonar
Hipertensión pulmonar secundaria a enfermedad cardiaca izquierda
Hipertensión pulmonar debida a enfermedad pulmonar o hipoxia
Hipertensión pulmonar debida a obstrucción arterial
Hipertensión pulmonar multifactorial o mecanismo no conocido
En los pacientes con SSc, la forma más frecuente es el tipo I, aunque también pueden presentar formas mixtas de la enfermedad2.
La definición fenotípica de hipertensión pulmonar que afecta a cada paciente sigue siendo un proceso difícil debido a la tendencia de dicha afección a presentar múltiples etiologías en el contexto de la SSc. Las comorbilidades de hipertensión pulmonar y fibrosis pulmonar están presentes en 25 a 50% de los pacientes, especialmente en aquellos con enfermedad difusa. Estos pacientes tienen un peor pronóstico y una mala respuesta al tratamiento. La enfermedad pulmonar venooclusiva se caracteriza por una obstrucción difusa de las venas pulmonares pequenas, y se puede encontrar en el 61% de los pacientes con SSc-HTP. Esta afección también se asocia con una menor supervivencia y un mayor riesgo de edema pulmonar después de iniciar el tratamiento. Adicionalmente, el compromiso cardíaco dado por fibrosis miocárdica puede ser una causa de SSc-HTP, especialmente de tipo II, al generar disfunción del ventrículo izquierdo3.
El tratamiento farmacológico de la SSc- HTP ha presentado grandes avances en los últimos anos, especialmente con el desarrollo de nuevos medicamentos para ser administrados por vía oral y el beneficio de las terapias combinadas con diferentes mecanismos de acción2. Los tratamientos se basan en la inducción de vasodilatación, y para ello se han utilizado los siguientes mecanismos de acción: antagonistas de los receptores de la endotelina-1 (bosentán, ambrisentán, macitentán, etc.), inhibidores de la fosfodiesterasa (sildenafil, tadalafil), inhibidores de la guanilato ciclasa (riociguat), y análogos de la prostaciclina (epoprostenol, treprostinil)1.
El objetivo de esta revisión sistemática de la literatura es evaluar la eficacia de los diferentes tratamientos farmacológicos disponibles para el manejo de la SSc-HTP.
Materiales y métodos
Criterios de selección
Tipos de estudios: ensayos clínicos controlados aleatorizados, ensayos clínicos controlados no aleatorizados, y estudios observacionales que incluyen estudios de cohortes y de casos y controles.
Se excluyeron los estudios observacionales sin grupo de comparación, los estudios que no evaluaron los desenlaces propuestos, las publicaciones duplicadas, los estudios en curso, y los estudios sin suficiente información para extraer resultados de interés para los pacientes con SSc.
Población
Se incluyeron los estudios con pacientes adultos (mayores de 18 anos) con diagnóstico de SSc-HTP que utilizaron cualquiera de los tratamientos farmacológicos disponibles.
Tipos de intervención
Estudios en los que algunos de los tratamientos disponibles para la SSc-HTP (análogos de la prostaciclina, antagonistas de los receptores de la endotelina, inhibidores de la fosfodiesterasa y estimuladores de la guanilato ciclasa soluble) se compararon con placebo o con un medicamento de otro grupo que se utiliza para tratar la SSc-HTP.
Desenlaces
Los desenlaces primarios incluidos en el estudio fueron la capacidad de ejercicio medida con la prueba de caminata de seis minutos, la funcionalidad medida con la clase funcional de la OMS o con la clasificación funcional de la NYHA, y el puntaje de disnea de Borg. Los desenlaces secundarios incluyeron mediciones hemodinámicas cardiopulmonares, niveles de NT-pro BNP, mortalidad y efectos adversos.
Métodos de búsqueda
La búsqueda de publicaciones se realizó en las siguientes bases de datos electrónicas científicas: Registro Central Cochrane de Ensayos Controlados (CENTRAL) (Cochrane Central Register of Controlled Trials), MEDLINE y EMBASE. La búsqueda se llevó a cabo entre enero de 1966 y abril de 2019.
Los términos de búsqueda incluidos fueron: hypertension, pulmonary (meSH) ORpulmonary hypertension ORpulmonary arterial hypertension AND scleroderma, systemic [meSH] OR systemic sclerosis AND treatment OR therapeutics [MeSH] OR bosentan OR ambrisentan OR macitentan OR sitaxsentan OR sildenafil OR tadalafil OR vardenafil OR riociguat OR selexipag OR epopros-tenol OR iloprost OR treprostinil OR beraprost.
Recolección y análisis de los datos
Selección de los estudios
Los estudios seleccionados fueron evaluados por dos investigadores. Se obtuvieron los siguientes datos: detalles de la publicación (título, primer autor, fecha de publicación), diseño del estudio, detalles de los participantes en el estudio (número de pacientes incluidos, características demográficas), tipo de intervención, tipo de comparador, tiempo de seguimiento, y desenlaces con su correspondiente medición del efecto.
Evaluación de la calidad y del riesgo de sesgo La calidad metodológica de los estudios se evaluó basándose en el tipo de diseño del estudio, siguiendo el Manual Cochrane de Revisiones Sistemáticas de Intervenciones5, incluyendo el riesgo de sesgo. En los estudios de cohortes y de casos y controles se aplicó la lista de verificación de la Red Escocesa Intercolegiada de Guías (Scottish Intercollegiate Guidelines Network, SIGN) para la evaluación metodológica6.
Medición de los efectos del tratamiento
Para la medición de los efectos del tratamiento farmacológico se presentó un análisis descriptivo y exploratorio de cada variable cuantitativa mediante medidas de tendencia central y dispersión, y para las variables cualitativas mediante medidas de frecuencia, incluyendo gráficos descriptivos. Finalmente, con base en los datos disponibles, se desarrolló un metaanálisis cuando fue posible.
Resultados
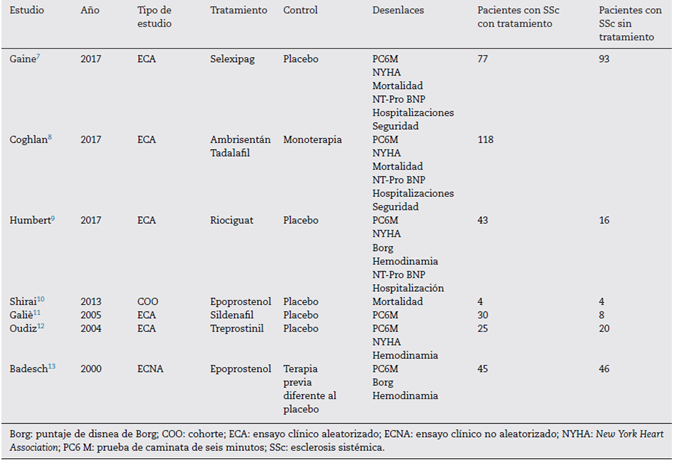
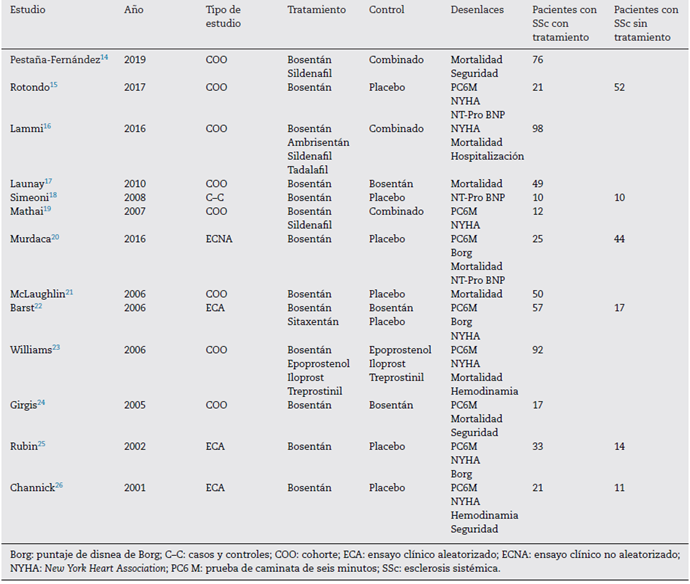
Durante la búsqueda inicial se encontraron en total 870 estudios. De éstos, se seleccionaron 195 basándose en el título; y luego se seleccionaron 120 basándose en los resúmenes para su lectura completa. Finalmente se incluyeron 20 estudios, ya que cumplieron con los criterios de inclusión (fig. 1). La definición de hipertensión arterial pulmonar ha sido cambiada a partir del año 2019, reduciendo el valor normal de la presión pulmonar mediana a 20 mmHg; sin embargo, todos los estudios utilizaron el punto de corte anterior de 25 mmHg. De los 20 estudios, ocho fueron ensayos clínicos aleatorizados, dos fueron ensayos clínicos no aleatorizados, nueve cohortes y un estudio de casos y controles. De estos, 13 estudios incluyeron al bosentán como monoterapia o en combinación. En las tablas 1 y 2 se describe un resumen de las características de los estudios.
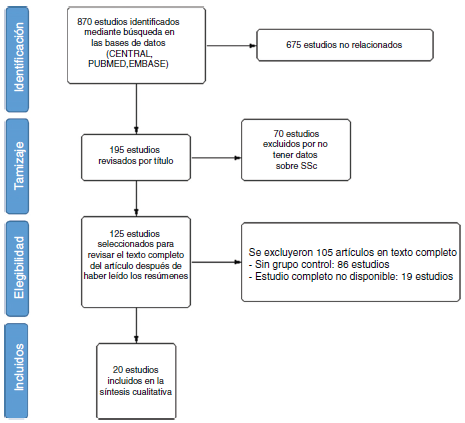
Después de esta búsqueda y análisis de los datos, se realizó una búsqueda activa específicamente para macitentán, vardenafil y beraprost, sin encontrar ninguna referencia adicional.
La evaluación de la calidad de la evidencia encontró que, en general, los ensayos clínicos tienen una adecuada calidad global. El riesgo de sesgo en los ensayos clínicos se presenta en las figuras 2 y 3. El estudio con más limitaciones fue el realizado por Badesch et al.13 que utiliza epoprostenol. Es importante que el otro estudio que utiliza este medicamento es de baja calidad10, como se mostrará más adelante. Los sesgos más frecuentes en los estudios se relacionaron con el cegamiento de los participantes, el personal y los investigadores.

Figura 2 Gráfico del riesgo de sesgo: revisa los dictámenes de los autores sobre cada elemento de riesgo de sesgo presentado como porcentajes en todos los estudios de ensayos clínicos incluidos.
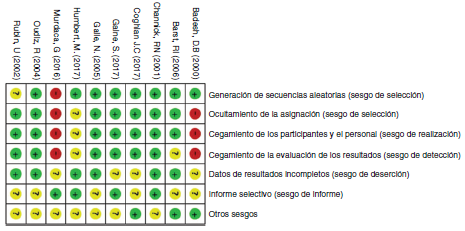
Figura 3 Resumen del riesgo de sesgo: revisa los dictámenes de los autores sobre cada elemento de riesgo de sesgo para cada estudio de ensayo clínico incluido.
La evaluación de la calidad de los estudios de cohortes y de casos y controles utilizando la lista de verificación SIGN específica para cada tipo de estudio se presenta en la tabla 3 y en la tabla 4, respectivamente. En general, la calidad de los estudios no fue la mejor, ya que pudieron presentarse varios sesgos, según la lista de verificación. Los sesgos más frecuentes fueron la falta de datos relacionados con la deserción de los participantes, el análisis de los datos de los participantes que desertaron y el cegamiento durante el análisis de los resultados.
Tabla 3 Evaluación de la calidad de la evidencia de los estudios de cohortes utilizando la lista de verificación SIGN
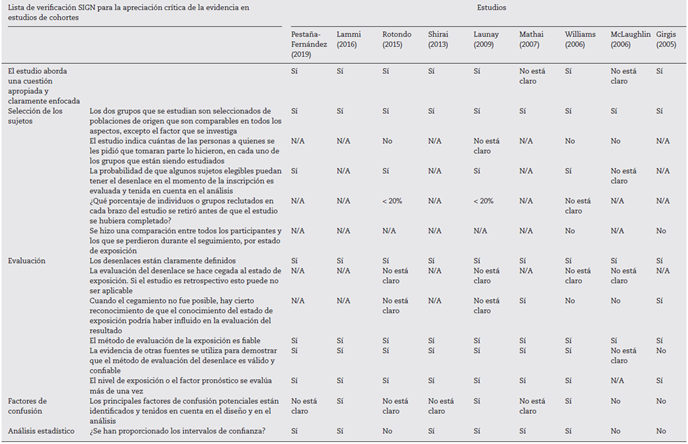
Tabla 4 Evaluación de la calidad de la evidencia de los estudios de casos y controles utilizando la lista de verificación SIGN

Los estudios que no incluyeron el bosentán como tratamiento son en su mayoría ensayos clínicos. Se incluyeron en total 342 pacientes con SSc-HTP con tratamiento, y 187 pacientes con placebo. A excepción de dos estudios, todos utilizaron medicamentos diferentes. Los dos estudios que usaron el epoprostenol como tratamiento10,13 no pudieron ser utilizados en el análisis posterior debido al tipo de estudio y a la cantidad limitada de pacientes incluidos. Dadas estas características de los estudios, no fue posible realizar un metaanálisis en el grupo que no utilizó el bosentán.
En cuanto a los resultados en pacientes con SSc, el xelexipag7 redujo el riesgo de eventos con base en un desenlace compuesto (incluyendo mortalidad, cambios en la clase funcional, hospitalizaciones y eventos adversos, requerimiento de trasplantes de pulmón, requerimiento de septostomía auricular con balón, y uso de prostanoides intravenosos), en comparación con el placebo (HR 0,59; IC 95% 0,41-0,85). Los pacientes seleccionados en el análisis del estudio formaron parte del ensayo GRIPHON, y fueron agrupados con aquellos con una etiología autoinmune de la HTP, incluyendo a los pacientes con SSc, lupus eritematoso sistémico, enfermedad mixta del tejido conectivo, e indiferenciada.
El riociguat9 a una dosis de 2,5 mg/día, mejoró la distancia en la prueba de caminata de seis minutos (18 ± 51 m), comparado con el placebo (-8 ± 110 m) después de 12 meses durante el ensayo PATENT-1, con pacientes que presentaron diferentes enfermedades autoinmunes. El efecto fue menor en los pacientes con SSc, en quienes la mejoría fue de 4 ±47 m, aunque el grupo control sin tratamiento presentó un mayor deterioro de -37 ± 120 m. Este beneficio continuó hasta los dos años de seguimiento, de acuerdo con los resultados del ensayo PATENT-2. Además, las variables hemodinámicas tales como la resistencia vascular pulmonar mejoraron, los niveles séricos de NT-pro BNP se redujeron y la clase funcional de la NYHA mejoró o se estabilizó en el 97% de los pacientes, siendo 22% mayor en comparación con el grupo control. No se encontraron diferencias en la mortalidad a dos años en comparación con los pacientes con HTP no autoinmune.
El sildenafil11 administrado a cualquier dosis mejoró la distancia en la prueba de caminata de seis minutos en 45-50 m (p = 0,001), redujo las presiones pulmonares en 2-4 mmHg de acuerdo con la dosis administrada (p > 0,05), y mejoró la clase funcional de la NYHA en un nivel en 20-30% de los pacientes, en comparación con el placebo, de acuerdo a la dosis administrada (IC 95% 16-42%; p > 0,05). Los eventos adversos tales como sofoco, diarrea y otros síntomas gastrointestinales aumentaron.
El treprostinil12 mejoró las variables hemodinámicas, incluyendo el índice cardíaco y la resistencia pulmonar (p = 0,006), mejoró la distancia promedio en la caminata de seis minutos a 25 m (p = 0,05) con un efecto dependiente de la dosis, siendo aquellos pacientes que recibieron más de 9 ng/kg/min los que presentaron mejorías mayores. Los puntajes de disnea mejoraron en comparación con el placebo después de 12 semanas de tratamiento. Se identificaron más efectos colaterales tales como reacciones en el sitio de la inyección y aquellos relacionados con el uso de prostanoides; sin embargo, los pacientes tuvieron una tolerancia adecuada en la mayoría de los casos, sin tener que suspender el tratamiento.
El uso del epoprostenol en el ensayo clínico no aleatorizado13 aumentó la distancia promedio de caminata de seis minutos en 46 m, en comparación con una reducción media de -48 m en el grupo control que utilizó la terapia convencional, después de 12 semanas de tratamiento. La diferencia de 108 metros fue significativa (IC 95% 58-180 m; p = 0,001). Se encontró una reducción de las presiones pulmonares de 6 mmHg, así como de las resistencias vasculares pulmonares; 21 pacientes del grupo de tratamiento presentaron mejoría en la clase funcional de la NYHA, mientras que ningún paciente del grupo control mejoró. El fenómeno de Raynaud y las úlceras digitales también mejoraron en los pacientes que recibieron epoprostenol. El estudio de casos y controles10 no permite sacar conclusiones adicionales debido a la muestra limitada de cuatro pacientes, aunque refiere una mejoría en la mortalidad.
La terapia combinada se evaluó con los pacientes del ensayo AMBITION8, en el que se comparó el tratamiento combinado de ambrisentán más tadalafil frente a la monoterapia con los mismos agentes. Los desenlaces primarios, incluyendo el fracaso terapéutico (definido como un empeoramiento de la clase funcional, de la prueba de caminata de seis minutos, el uso de prostanoides, el requerimiento de trasplante o septostomía ventricular) se produjeron en menor proporción en los pacientes que recibieron terapia combinada (20%), en comparación con los que recibieron monoterapia (41%) (HR 0,44; IC 95% 0,22-0,89). Considerando los desenlaces secundarios, se encontró una reducción de los niveles NT-pro BNP y un incremento de 404 ± 12 m en la prueba de caminata de seis minutos. Con respecto a los eventos adversos, se desarrolló edema periférico en 44% de los pacientes con terapia combinada, en comparación con el 26% de los pacientes que recibieron monoterapia con ambrisentán y el 33% de los pacientes con monoterapia con tadalafil. No se encontraron diferencias en el desarrollo de cefalea, anemia o síncope.
La mayoría de los estudios que incluyeron el bosentán fueron de cohortes, seguidos de tres ensayos clínicos controlados (dos evaluaron al bosentan vs. placebo, y uno también evaluó el sitaxsentán), y un estudio de casos y controles. Se incluyeron en total 504 pacientes con SSc-HTP que recibieron tratamiento, y 148 pacientes sin tratamiento para los grupos de control.
El ensayo clínico piloto26 agrupó a 32 pacientes con SSc con funcionalidad clase III de la NYHA, aleatorizados en una relación 2:1 para el bosentán 125 mg cada 12 horas o placebo. Después de 12 semanas de tratamiento, se encontró un incremento de 76 m en la prueba de caminata de seis minutos (IC 95% 12-139 m; p = 0,02). La mejoría se mantuvo después de 20 semanas de tratamiento. También se encontró una mejoría en los parámetros hemodinámicos, incluyendo el índice cardíaco y una reducción de las resistencias vasculares (p = 0,002), y en los desenlaces preestablecidos (puntaje de disnea de Borg, clase funcional de la NYHA, y suspensión de la medicación debida a empeoramiento clínico). Sin embargo, estos datos se obtuvieron de todo el grupo de pacientes, y no se hizo un análisis de estratificación para identificar las mejorías únicamente en los pacientes con SSc.
El ensayo BREATHE-125 comparó el bosentán vs. placebo en pacientes con HTP, incluyendo a aquellos con SSc. Se encontró una mejoría en la prueba de caminata de seis minutos (44 m; IC 95% 21-67 m; p > 0,001), el puntaje de disnea de Borg y la clase funcional de la OMS. Basándose en el análisis de subgrupos, los autores consideraron que el bosentán puede tener un efecto preventivo en los pacientes con SSc, considerando una disminución de la capacidad de ejercicio, con una mejora de 3 m en comparación con el placebo (reducción de 40 m) utilizando la prueba de caminata de seis minutos. No se encontraron diferencias significativas en la mortalidad. El perfil de seguridad mostró que la presencia de perfiles bioquímicos hepáticos anormales era dependiente de la dosis.
Los seguimientos durante tres años de los pacientes incluidos en estos ensayos se compararon con controles históricos, mostrando una supervivencia a un año del 85%, y una tasa de supervivencia a dos años del 70% con la monoterapia con bosentán. La supervivencia de los pacientes con bosentán vs. epoprostenol fue del 82% durante el primer año de tratamiento, y del 67% durante el segundo año, en 30% de los pacientes. Los seguimientos también mostraron que la supervivencia fue más baja en los pacientes con SSc, comparada con la de aquellos con HTP idiopática21.
El estudio que evaluó el uso del sitaxsentán vs. placebo y vs. bosentán22 en la distancia de la caminata de seis minutos utilizó un brazo abierto sin cegamiento para el bosentán. Se incluyeron pacientes con HTP de diferentes etiologías (30% de los pacientes con HTP estaban asociados a enfermedades del tejido conectivo, sin especificar cuántos pacientes tenían SSc). En todos los pacientes se observó una mejoría de 31,4 m con una dosis de 100 mg (p = 0,03); de 24,2m con una dosis de 50?mg (p = 0,07), y de 29,5 m con el bosentán (p = 0,05), comparada con placebo. Adicionalmente, el sitaxentán mejoró la clase funcional de la OMS (p = 0,04).
Entre los estudios no aleatorizados, se encontró una cohorte retrospectiva del registro español RESCLE14. De 1.817 pacientes con SSc, 76 tenían SSc-HTP, y estaban recibiendo monoterapia con inhibidores de la fosfodiesterasa o con antagonistas de los receptores de la endotelina como terapia inicial combinada, o como terapia secuencial combinada. Ellos compararon las tasas de mortalidad entre los grupos a uno, tres y cinco años. El grupo de la combinación secuencial tuvo la mortalidad más baja (HR 0,11; IC 95% 0,03-0,5; p = 0,004). La supervivencia a un año fue del 78% para la monoterapia, del 94% para la terapia combinada inicial, y del 95% para la terapia secuencial; la supervivencia a tres años fue del 40% para la monoterapia, del 51% para la terapia inicial combinada, y del 81% para la terapia secuencial; la supervivencia a cinco años fue del 31% para la monoterapia, del 34% para la terapia inicial combinada, y del 56% para la terapia secuencial (p = 0,007). No se encontraron diferencias significativas entre los grupos con respecto al desarrollo de eventos adversos.
El estudio realizado por Launay et al.17 también analizó la mortalidad con el uso de la terapia combinada con inhibidores de la fosfodiesterasa. Se compararon los datos de supervivencia con una cohorte del mismo grupo de estudio que fue publicada anteriormente. Las tasas de supervivencia a uno, tres y cinco años fueron del 92%, 89% y 79%, respectivamente con el bosentán; y del 80%, 56% y 51% para los pacientes con SSc-HTP (p = 0,001). Otro estudio que incluyó un análisis de la mortalidad23 mostró diferencias entre el grupo control y aquellos pacientes que utilizaban el bosentán. Sin embargo, el grupo control estaba utilizando prostaciclinas, y los pacientes tenían una fibrosis pulmonar más severa, y una peor capacidad pulmonar, lo que hizo que los resultados no fueran fiables, ya que estas afecciones no fueron consideradas durante el análisis.
Los demás estudios de casos y controles y de cohortes reunieron a 449 pacientes y mostraron desenlaces variables. Dos estudios en pacientes con SSc-HTP evaluaron los niveles de NT-pro BNP como el principal resultado17,18, sin descubrir una reducción significativa después del primer o segundo anno utilizando el bosentán. No se encontró una relación con el cambio significativo en la clase funcional de la NYHA (p = 0,01) y la mejoría en la distancia de la caminata de seis minutos (p = 0,04). El otro estudio con menos pacientes encontró una reducción del NT-pro BNP después de siete meses de tratamiento, pero no fue estadísticamente significativa (p = 0,6).
Un tercer estudio20 evaluó los niveles de NT-pro BNP en pacientes con SSc que desarrollaron úlceras digitales y fueron tratados con bosentán. También evaluó el desarrollo de HTP mediante estimación ecocardiográfica, encontrando una reducción de la presión pulmonar media. Ningún paciente del grupo que recibió bosentán desarrolló HTP, comparado con aquellos del grupo control en el que siete pacientes desarrollaron dicha afección. Los pacientes del grupo de bosentán lograron una reducción de los valores de la presión pulmonar, mientras que los del grupo control presentaron un aumento de estos valores (p = 0,001). Los puntajes de disnea de Borg mejoraron en el grupo de bosentán (p = 0,001) y los valores de NT-pro BNP disminuyeron en comparación con los iniciales, en contraste con el grupo control (p = 0,004).
Una cohorte de 98 pacientes16 de la base de datos del registro PHAROS comparó el efecto de adicionar inhibidores de la fosfodiesterasa a la monoterapia de antagonistas de los receptores de endotelina con bosentán o ambrisentan. El desenlace del tiempo transcurrido hasta el empeoramiento clínico se definió como un aumento de los síntomas, muertes, hospitalizaciones o de la necesidad de prostanoides. Elempeoramiento clínico se desarrolló más temprano en el grupo de pacientes con monoterapia, en comparación con aquellos que recibieron terapia combinada o inhibidores de la fosfodiesterasa. El análisis multivariado mostró que el uso inicial de la monoterapia con antagonistas de los receptores de endotelina se asoció con un tiempo más corto hasta el empeoramiento clínico, en comparación con la terapia combinada o con los inhibidores de la fosfodiesterasa (HR 2,63; IC 95% 1,28-5,56; p = 0,009). No se encontraron diferencias significativas entre los grupos al inicio del seguimiento.
Otro estudio analizó la combinación de antagonistas de los receptores de la endotelina con inhibidores de la fosfodiesterasa en pacientes con HTP en quienes fracasó la monoterapia con bosentán. Se encontró que en los pacientes con SSc, la mejoría de la clase funcional de la NYHA y de la distancia de la caminata de seis minutos fue inferior. Esta misma comparación entre los grupos (SSc-HTP vs. HTP idiopática) utilizando la monoterapia con bosentán24 encontró los mismos resultados, con mejores desenlaces de respuesta en los pacientes con HTP idiopática vs. aquellos con SSc, respecto a la clase funcional de la OMS.
Los investigadores intentaron desarrollar un metaanálisis, especialmente para aquellos estudios que utilizaron el bosentán. Sin embargo, la falta de datos específicos y la heterogeneidad en los grupos de comparación no permitió hacerlo para resultados tales como los puntajes de disnea o la clase funcional de la NYHA.
Dos ensayos clínicos del bosentán vs. placebo evaluaron la prueba de caminata de seis minutos como el desenlace principal. En el estudio de Rubin et al.25 los datos pudieron haber sido extraídos en forma inexacta como un gráfico. Sin embargo, en el estudio realizado por Channick et al.26 no se suministraron datos específicos para los pacientes con SSc, lo que imposibilitó el desarrollo de un metaanálisis. En los estudios observacionales, no se hizo un metaanálisis respecto a la distancia de caminata de seis minutos porque: 1) un estudio no utilizó un grupo control como comparación15, y 2) otro estudio comparó los resultados con los de un grupo de pacientes con HTP idiopática19,23.
Discusión
Después de la revisión sistemática de la literatura, se encontró que hay evidencia disponible, derivada de ensayos para el tratamiento de la HTP que incluyen pacientes con SSc. Estos ensayos muestran un efecto beneficioso con el uso de diferentes agentes farmacológicos, representado especialmente por la mejora de la distancia de caminata de seis minutos, que fue la prueba utilizada más frecuentemente durante el seguimiento de los pacientes. Los efectos positivos en la prueba de caminata de seis minutos se observaron con el uso de antagonistas de los receptores de endotelina tales como el ambrisentán y el bosentán (este último con la mayor cantidad de estudios), prostanoides, inhibidores de la fosfodiesterasa como el sildenafil, y el riociguat que también presenta mejoras en otros desenlaces incluyendo la clase funcional de la NYHA y el puntaje de disnea de Borg. El desenlace de mortalidad también fue satisfactorio en varios estudios, ya que las tasas de supervivencia presentaron un aumento en comparación con las cohortes históricas e incluso mostraron tasas similares a las de los pacientes con diferentes etiologías de HTP con mejor pronóstico, tales como las de la HTP idiopática. Asimismo, varias mediciones hemodinámicas y marcadores bioquímicos como el NT-pro BNP mostraron mejorías durante el seguimiento.
A pesar de estos hallazgos, aún existen varias dificultades e interrogantes por resolver sobre este tema. Se requieren más datos para resolver las siguientes incógnitas: identificar la mejor terapia para este grupo de pacientes, saber si estos pacientes se benefician más con una terapia combinada temprana, si alguno de los agentes farmacológicos es superior a otros, y el impacto de tratamientos no farmacológicos tales como la rehabilitación. También hay datos limitados sobre pacientes con patologías pulmonares concomitantes, que son frecuentes en los tienen SSc.
Los beneficios presentados por la evidencia disponible deben ser evaluados con herramientas que puedan demostrar también mejoras directas de los síntomas y la calidad de vida, especialmente considerando que el resultado más frecuente evaluado en los estudios fue la prueba de caminata de seis minutos, que tiene alguna relación con la mortalidad en las mediciones iniciales, pero su variación con respecto a la medición inicial tiene un significado pronóstico limitado. Aun así, otros resultados hemodinámicos y bioquímicos tienen mayor valor pronóstico para la mortalidad, pero no fueron utilizados con frecuencia en los estudios incluidos.
La calidad global de la evidencia para los estudios incluidos fue moderada para los ensayos clínicos, según la clasificación GRADE, y en algunos casos fue alta7,11,12,16, ya que se derivaron de los principales ensayos sobre tratamiento farmacológico. La clasificación GRADE para los ensayos no aleatorizados fue baja, y en algunos casos muy baja debido a su riesgo de sesgo.
En cuanto al desarrollo de un metaanálisis de los estudios seleccionados, no fue posible en ningún caso debido a la heterogeneidad del diseño, al número de los distintos agentes farmacológicos estudiados, a las poblaciones de comparación, a las diferencias en los tiempos de seguimiento para la medición de los desenlaces, y a la ausencia de datos específicos para los pacientes con SSc.
Conclusión
La evidencia disponible sobre el tratamiento farmacológico de la SSc-HTP muestra que el uso de medicamentos aprobados se asocia a una mejora de los desenlaces clínicos y no clínicos, favoreciendo su implementación en los esquemas terapéuticos para esta enfermedad. La mayoría de los estudios sugieren el uso de terapias combinadas con múltiples agentes farmacológicos para lograr una mejor respuesta. Sin embargo, la evidencia disponible sigue siendo limitada para los pacientes con SSc, y se requieren investigaciones futuras para desarrollar recomendaciones específicas con respecto al mejor tratamiento farmacológico y la mejor terapia combinada.
REFERENCIAS
1. Allanore YSimms R, Distler O, Trojanowska M, Pope J, Denton C, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1. [ Links ]
2. Denton C, Wells A, Coghlan J. Major lung complications of systemic sclerosis. Nat Rev Rheumatol. 2018;14:511-27. [ Links ]
3. Launay D, Sobanski V, Hachulla E, Humbert M. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev. 2017;26:170056. [ Links ]
4. Sundaram S, Chung L. An update on systemic sclerosis-associated pulmonary arterial hypertension: a review of the current literature. Curr Rheumatol Rep. 2018;20. [ Links ]
5. Higgins JPT, Green S, editors. Cochrane handbook for systematic reviews of interventions, version 5.1.0. The Cochrane Collaboration; 2011. Available from: Available from: www.handbook.cochrane.org [updated March 2011]. [ Links ]
6. Scottish Intercollegiate Guidelines Network. Critical appraisal: notes and checklists. https://www.sign.ac.uk/checklists-and-notes.html [Internet]. [ Links ]
7. Gaine S, Chin K, Coghlan G, Channick R, Di Scala L, Galiè N, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J. 2017;50:1602493. [ Links ]
8. Coghlan J, Galiè N, Barberà J, Frost A, Ghofrani H, Hoeper M, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis. 2016;76:1219-27. [ Links ]
9. Humbert M, Coghla J, Ghofrani H, Grimminger F, He J, Riemekasten G, et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis. 2016;76:422-6. [ Links ]
10. Shirai Y, Yasuoka H, Takeuchi T, Satoh T, Kuwana M. Intravenous epoprostenol treatment of patients with connective tissue disease and pulmonary arterial hypertension at a single center. Mod Rheumatol. 2013;23:1211-20. [ Links ]
11. Galiè N, Ghofran H, Torbicki A, Barst R, Rubin L, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Eng J Med. 2005;353:2148-57. [ Links ]
12. Oudiz R, Schilz R, Barst R, Galié N, Rich S, Rubin L, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126:420-7. [ Links ]
13. Badesch D, Tapson V, McGoon M, Brundage B, Rubin L, Wigley F, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. Ann Intern Med. 2000;132:425-34. [ Links ]
14. Pestaiia-Fernández M, Rubio-Rivas M, Tolosa-Vilella C, Guillén-Del-Castillo A, Freire M, Vargas-Hitos J, et al. Longterm efficacy and safety of monotherapy versus combination therapy in systemic sclerosis-associated pulmonary arterial hypertension: a retrospective RESCLE Registry Study. J Rheumatol. 2019, http://dx.doi.org/10.3899/jrheum.180595. [ Links ]
15. Rotondo C, Praino E, Nivuori M, di Serio F, Lapadula G, Iannone F. No changes in N-terminal pro-brain natriuretic peptide in a longitudinal cohort of patients with systemic sclerosis-associated pulmonary arterial hypertension on therapy with bosentan. Int J Rheum Dis. 2015;20:90-6. [ Links ]
16. Lammi M, Mathai S, Saketkoo L, Domsic R, Bojanowski C, Furst D, et al. Association between initial oral therapy and outcomes in systemic sclerosis-related pulmonary arterial hypertension. Arthritis Rheumatol. 2016;68:740-8. [ Links ]
17. Launay D, Sitbon O, Le Pavec J, Savale L, Tcherakian C, Yaici A, et al. Long-term outcome of systemic sclerosis-associated pulmonary arterial hypertension treated with bosentan as first-line monotherapy followed or not by the addition of prostanoids or sildenafil. Rheumatology. 2009;49:490-500. [ Links ]
18. Simeoni S, Lippi G, Puccetti A, Montagnana M, Tinazzi E, Prati D, et al. N-terminal pro-BNP in sclerodermic patients on bosentan therapy for PAH. Rheumatol Int. 2007;28:657-60. [ Links ]
19. Mathai S, Girgis R, Fisher M, Champion H, Housten-Harris T, Zaiman A, et al. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Resp J. 2007;29:469-75. [ Links ]
20. Murdaca G, Lantieri F, Puppo F, Bezante G, Balbi M. Beneficial effects of long-term treatment with bosentan on the development of pulmonary arterial hypertension in patients with systemic sclerosis. J Int Med Res. 2016;44 1 suppl.:85-9. [ Links ]
21. McLaughlin V. Survival in patients with pulmonary arterial hypertension treated with first-line bosentan. Eur J Clin Invest. 2006;36 Suppl. 3:10-5. [ Links ]
22. Barst R, Langleben D, Badesch D, Frost A, Lawrence E, Shapiro S, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47:2049-56. [ Links ]
23. Williams M, Das C, Handler CE, Akram MR, Davar J, Denton CP, et al. Systemic sclerosis associated pulmonary hypertension: improved survival in the current era. Heart. 2006;92:926-32. [ Links ]
24. Girgis R, Mathai S, Krishnan J, Wigley F Hassoun P. Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderm spectrum of diseases. J Heart Lung Transplant. 2005;24:1626-31. [ Links ]
25. Rubin L, Badesch D, Barst R, Galiè N, Black C, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Eng J Med. 2002;346:896-903. [ Links ]
26. Channick R, Simonneau G, Sitbon O, Robbins I, Frost A, Tapson V, et al. Effects of the dual endothelin-receptor antagonist Bosentan in patients with pulmonary hypertension: a randomized placebo-controlled study. Lancet. 2001;358:1119-23. [ Links ]
Recibido: 26 de Noviembre de 2019; Aprobado: 19 de Mayo de 2020
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
