











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Las fitoalexinas son metabolitos secundarios (actualmente conocidos como especializados) producidos por las plantas para protegerse contra patógenos, metales pesados y radiación ultravioleta; se constituyen como compuestos cruciales de los mecanismos de defensa de las plantas [1]. Entre las fitoalexinas liberadas por las cruciferas (Brassicaceae), la brassinina tiene un papel fundamental, no solo como respuesta defensiva, sino también en la vía biosintética de aquellas derivadas de ʟ-triptófano, dado que es un precursor importante de varias fitoalexinas, por ejemplo, ciclobrassinina, brasilexina, espirobrassinina, entre otras [2]. Este tipo de compuestos ha generado gran importancia por su actividad biológica frente a patógenos como Alternaria brasiccola, Botrytis cinérea, Leptosphaeria maculans, Rhizoctonia solani, entre otros [3], [4]. Recientemente, el grupo Integrado de Investigaciones en Química Bioorgánica (InQuiBio), de la Universidad Militar Nueva Granada, ha orientado sus investigaciones hacia la síntesis de análogos de fitoalexinas indólicas, utilizando 2-aminoácidos convencionales como precursores sintéticos, los cuales puedan comportarse como bioisósteros de fitoalexinas heterocíclicas para ser usados como agentes antifúngicos contra Fusarium oxysporum [ 5]-[8]. Estos antecedentes han inspirado la búsqueda de nuevas moléculas bioactivas que tengan núcleos estructurales interesantes, de los cuales ya se cuenta con un conocimiento previo sobre su potencial actividad biológica.
Dando continuidad a nuestro propósito, hemos estudiado varios grupos funcionales; buscando identificar su relevancia en la actividad biológica de las fitoalexinas. Particularmente, hemos iniciado la exploración de grupos funcionales nitrogenados, como las bases de Schiff y en especial las amidas N-alquilsustituidas, las cuales han sido identificadas como farmacóforos [9]-[14]. Dada su relevancia, la síntesis de amidas se ha convertido en un tema de investigación relevante en la síntesis química, dirigido al desarrollo de moléculas biológicamente activas con potenciales actividades anticancerígenas, antibacteriales, analgésicas, antiinflamatorias y antifúngicas [15]-[21]. Las metodologías tradicionales para la síntesis de amidas involucran el uso de aminas primarias y secundarias, y derivados de ácidos carboxílicos activados [22], por ejemplo, cloruros de acilo o anhídridos. Sin embargo, el uso de agentes de acoplamiento C-N, a partir de la activación del grupo ácido carboxílico, como la formación de ésteres activados con carbodiimidas, con EDC y DCC, la formación de cloruro de ácido con cloruro de tionilo y cloruro de oxalilo como reactivos preferidos, y CDI, cloruro de pivaloílo (PivCl), cloroformiato de isobutilo (IBCF) y anhídrido de ácido n-propanofosfónico (T3P) para la preparación de anhídridos mixtos [23], resulta tóxico, costoso y genera gran cantidad de residuos nocivos para el medioambiente. Algunas de estas metodologías suelen presentar dificultades debido a los tiempos largos de reacción y bajos rendimientos, y dan lugar a grandes cantidades de subproductos que conllevan procedimientos de purificación difíciles [24]. Por tal motivo, en los últimos años se han planteado nuevas estrategias que involucran el uso de catalizadores ácidos de Lewis o empleando catálisis básica mediante aminas [25]-[27]. Por otro lado, la utilización de la irradiación por microondas (MW) ha permitido mejoras significativas en los porcentajes de rendimiento y cortos tiempos de reacción [28], [29], específicamente para la síntesis de N-acilendiaminas [30], [31], amidas monosustituidas [32], amidas N-heterocíclicas [33], entre otras.
Basándonos en los antecedentes descritos anteriormente y la estructura molecular de algunas fitoalexinas indólicas biológicamente activas, en este artículo se discute la síntesis asistida por irradiación de microondas, de amidas N-alquilsustituidas derivadas de 2-aminoácidos (i. e., L-triptófano, L-alanina, L-fenilalanina y L-tirosina); se evalúa el uso de tricloruro de aluminio (que actúa como catalizador) y el de 4-dimetilaminopiridina (con un rol como auxiliar nucleofílico). Se discuten las propuestas mecanísticas de cada método de reacción para la formación de amidas N-alquilsustituidas. Finalmente, los compuestos obtenidos fueron evaluados frente a Fusarium oxysporum en un medio suplementado; se determinó el valor del IC50 de cada compuesto, demostrando su actividad antifúngica a diferentes niveles, lo cual se interpretó en razón del efecto de la amida acíclica como bioisóstero de fitoalexinas heterocíclicas.
Materiales y métodos
Información general
Todos los reactivos y productos químicos se adquirieron comercialmente (Merck KGaA y/o Sigma-Aldrich) y fueron empleados sin purificación adicional. Todos los compuestos obtenidos fueron purificados mediante cromatografía flash empleando silica gel (Mesh 230-400) y como fase móvil éter de petróleo/acetato de etilo en proporción variable. La pureza de los compuestos se comprobó mediante cromatografía en capa fina realizada con láminas de aluminio Merck Silica Gel 60 F254. Las manchas se detectaron por su absorción bajo luz UV (254 nm) y relevado empleando yodo molecular, ninhidrina y sulfato cérico amónico. Los experimentos de RMN unidimensionales y bidimensionales se registraron en un espectrómetro Bruker Avance III HD Ascend de 400 MHz utilizando CDCl3 como disolvente con tetrametilsilano (TMS) como patrón interno a temperatura ambiente. Los desplazamientos químicos se indican en 5 (ppm) con respecto al TMS y las constantes de acoplamiento J se indican en hercios (Hz). Los experimentos de cromatografía líquida acoplada a espectrometría de masas (LC/MS) para corroborar la pureza de las muestras se realizaron en un espectrómetro LCMS 2020 (Shimadzu, Columbia, MD), que comprende un sistema de cromatografía líquida de alto rendimiento (HPLC) Prominence acoplado a un analizador de cuadrupolo simple con ionización por electrospray (ESI). Se utilizó una columna Synergi (150 x 4,6 mm, 4,0 um) para el análisis a 0,6 mL/min utilizando mezclas de acetonitrilo (A) y ácido fórmico al 1% v/v (B) en elución en gradiente. La ESI-MS se operó simultáneamente en los modos de iones positivos y negativos (barrido 100-2000 m/z), una temperatura de la línea de desolvatación a 250 °C, nitrógeno como gas nebulizador a 1,5 L/min, un gas de secado a 8 L/min, y un voltaje del detector a 1,4 kV, determinando el espectro de masas de cuatro compuestos seleccionados aleatoriamente. Se registraron datos de masa exacta a los compuestos 12, 14, 15 y 16, mediante mediciones por MS de alta resolución (HRMS) en un espectrómetro de masas micrOTOF-Q II (Bruker, Billerica, MA, EE. UU.). La ESI también se operó en los modos de iones positivos y negativos (barrido 100-2000 m/z), una temperatura de la línea de desolvatación a 250 °C, nitrógeno como gas nebulizador a 1,5 L/min, un gas de secado a 8 L/min, una energía cuadrupolar a 7,0 eV y una energía de colisión a 14 eV. La rotación óptica específica para cada compuesto se determinó con un polarímetro Jasco P-2000 (Jasco Co., Ltd., Mary's Court, PA, EE. UU.) en una celda de cuarzo (1,0 cm).
Síntesis de amidas N-alquilsustituidas
La síntesis de las amidas 1-16 se realizó empleando dos métodos bajo irradiación de microondas (MW): en el método A se empleó un ácido de Lewis como el tricloruro de aluminio (AlCl3) y en el método B se utilizó un auxiliar nucleofílico como la 4-dimetilaminopiridina (DAMP).
Método A. Síntesis de amidas con ácido de Lewis (AlCl3)
Las amidas se prepararon basándose en la metodología previamente reportada en la literatura con algunas modificaciones [34]. Los 2-aminoésteres se prepararon bajo irradiación por microondas (MW): se mezclaron los 2-aminoácidos (1 mmol) con TMSCl (4 mmol) y el alcohol respectivo (1 mL). La mezcla se calentó a 120 °C durante 20 min bajo irradiación de MW en atmósfera cerrada. Posteriormente, se dispuso la mezcla en un balón de fondo redondo y se concentró a presión reducida. El 2-aminoéster de alquilo, en forma de clorhidrato crudo, se dispuso en un tubo reactor del equipo de MW, se adicionó Et3N (4 mmol) y malonato de dietilo (DEM: 1 mmol), tolueno (1,5 mL), AlCl3 (0,2 mmol) y ácido acético (0,5 mmol). La mezcla de reacción se llevó a irradiación por MW a 150 °C por 20 min. Finalizado este tiempo, y al no detectar el reactivo de partida por CCD, el crudo de reacción se acidificó con HCl diluido hasta obtener un pH débilmente ácido (entre 5 y 6). A continuación, se realizó extracción con acetato de etilo (5 x 10 mL), se secó con sulfato de sodio anhidro y, posteriormente, se concentró bajo presión reducida. El crudo de reacción se purificó por cromatografía en columna flash, eluyendo con mezclas de éter de petróleo/ acetato de etilo de polaridad variable, para dar los productos 1-16. Los rendimientos de cada compuesto se muestran en la Figura 2.
Método B. Síntesis de amidas con DAMP
Los 2-aminoésteres se prepararon bajo irradiación por MW: se mezclaron los 2-aminoácidos (1 mmol) con TMSCl (4 mmol) y el alcohol respectivo (1 mL). La mezcla se calentó a 120 °C durante 20 min bajo irradiación de MW en atmósfera cerrada. La mezcla de reacción se concentró a presión reducida y se redisolvió en THF (1 mL). Por último, esta disolución se calentó a 140 °C durante 20 min bajo MW tras la adición de DEM (1 mmol) y DMAP (0,5 mmol). La mezcla de reacción se llevó a sequedad por destilación al vacío y el crudo se purificó por cromatografía en columna flash; se eluyó con mezclas éter de petróleo-acetato de etilo de polaridad variable, para obtener los productos 1-16. Los rendimientos de cada compuesto se muestran en la Figura 2 y los datos espectroscópicos de los compuestos se exponen a continuación.
N a -(3-etoxi-3-oxopropanoil)-L-triptofanato de metilo (1, C17H20N2O5)
Aceite amarillo; RMN 1H (400 MHz, CDCl3) δH 8,12 (s, 1H); 7,54 (d; J = 7,9 Hz; 1H); 7,35 (d; J = 8,1 Hz; 1H); 7,26 (s; 1H); 7,21-7,16 (m; 1H); 7,13-7,08 (m; 1H); 7,06 (d; J = 2,4 Hz; 1H); 4,93 (dt; J = 7,5; 5,6 Hz; 1H); 4,12 (q; J = 7,1 Hz; 2H); 3,69 (s; 3H); 3,34 (t; J = 4,9 Hz; 2H); 3,28 (d; J = 3,6 Hz; 2H); 1,61 (s; 3H); RMN 13C (100 MHz; CDCl3) δC 172,2; 169,0; 164,9; 136,2; 127,7; 123,0; 122,3; 119,8; 118,7; 111,4; 110,1; 77,5; 77,2; 76,8; 61,7; 53,2; 52,6; 41,5; 29,8; 27,7; 14,1; [α]25 D = +5,89° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-triptofanato de etilo (2, C18H22N2O5)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 8,27 (s; 1H); 7,55 (d; J = 7,9 Hz; 1H); 7,50 (d; J = 7,5 Hz; 1H); 7,33 (d; J = 8,1 Hz; 1H); 7,197,14 (m; 1H); 7,09 (t; J = 7,6 Hz; 1H); 7,04 (d; J = 2,4 Hz; 1H); 4,92 (dt; J = 7,6; 5,6 Hz; 1H); 4,14-4,07 (m; 4H); 3,34 (t; J = 5,7 Hz; 2H); 3,26 (d; J= 2,8 Hz; 2H); 1,21 (td; J = 7,1; 4,4 Hz; 6H); RMN 13C (100 MHz; CDCl3) δC 171,8; 168,9; 165,0; 136,2; 127,8; 123,0; 122,2; 119,6; 118,7; 111,3; 110,1; 61,7; 53,3; 41,5; 27,7; 14,2; [α]25 D = +9,55° ± 0,01 (c 0,6; MeOH).
N a (3-etoxi-3-oxopropanoil)-L-triptofanato de isopropilo (3, C19H24N2O5)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 8,79 (d; J = 7,9 Hz; 1H); 7,58-7,53 (m; 2H); 7,31 (d; J = 8,0 Hz; 1H); 7,19-7,08 (m; 1H); 7,00 (s; 1H); 4,92 (dq; J = 27,6; 6,3 Hz; 2H); 4,07 (q; J = 7,1 Hz; 1H); 3,31 (qd; J= 14,8; 5,8 Hz; 2H); 1,21-1,15 (m; 8H); RMN 13C (100 MHz; CDCl3) δC 172,3; 169,9; 167,0; 69,1; 48,5; 42,5; 21,7; 17,8; [α]25 D = +2,46° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-triptofanato de n-butilo (4, C20H26N2O5)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 8,69 (t; J = 3,5 Hz; 1H); 7,53 (d; J = 7,9 Hz; 2H); 7,47 (d; J = 7,5 Hz; 0H); 7,31 (d; J = 8,1 Hz; 1H); 7,15 (t; J= 7,5 Hz; 1H); 7,08 (t; J= 7,4 Hz; 1H); 7,00 (d; J= 2,3 Hz; 1H); 4,93 (q; J = 5,9 Hz; 1H); 4,14-4,00 (m; 4H); 3,32 (t; J = 5,8 Hz; 1H); 3,24 (d; J= 2,2 Hz; 2H); 1,58-1,48 (m; 3H); 1,37-1,23 (m; 3H); 1,19 (t; J = 7,1 Hz; 1H); 0,89 (q; J = 7,3 Hz; 4H); RMN 13C (100 MHz; CDCl3) δC 171,8; 168,8; 165,1; 136,2; 127,6; 123,1; 121,9; 119,4; 118,4; 111,4; 109,5; 65,4; 61,6; 53,3; 41,4; 30,4; 27,6; 19,0; 13,7; [α]25 D = +5,13° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-fenilalaninato de metilo (5, C15H19NO5)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 7,48 (d; J = 7,6 Hz; 1H); 7,33-7,10 (m; 5H); 4,87 (dt; J = 7,5; 6,0 Hz; 1H); 4,18 (q; J = 7,1 Hz; 2H); 3,72 (s; 3H); 3,29 (d; J = 2,7 Hz; 2H); 3,22-3,04 (m; 2H); 1,26 (t; J = 7,2 Hz; 4H); RMN 13C (100 MHz; CDCl3) δC 171,8; 168,9; 164,8; 135,9; 129,4; 128,7; 127,3; 61,8; 53,6; 52,5; 41,4; 37,9; 14,1; [α]25 D = +6,50° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-fenilalaninato de etilo (6, C16H21NO5)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 7,46 (d; J= 7,6 Hz; 1H); 7,36-7,09 (m; 4H); 4,85 (dt; J = 7,6; 6,1 Hz; 1H); 4,24-4,07 (m; 4H); 3,30 (d; J = 2,2 Hz; 1H); 3,14 (qd; J = 13,9; 6,1 Hz; 1H); 1,29-1,23 (m; 6H); RMN 13C (100 MHz; CDCl3) δC 171,3; 168,9; 164,8; 136,0; 129,5; 128,6; 127,2; 61,8; 61,7; 60,5; 53,6; 41,5; 38,0; 21,2; 14,1; [α]25 D = +1,50° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-fenilalaninato de isopropilo (7, C17H23NO5)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,60 (dd; J = 11,5; 7,7 Hz; 1H); 7,25 (p; J = 6,9 Hz; 3H); 7,14 (t; J = 6,0 Hz; 2H); 6,40 (d; J = 8,1 Hz; 1H); 5,03-4,93 (m; 1H); 4,79 (td; J = 21,4; 6,7 Hz; 1H); 4,09 (q; J = 7,1 Hz; 2H); 3,07 (q; 4H); 1,96 (s; 3H); 1,27-1,11 (m; 9H); RMN 13C (100 MHz; CDCl3) δC 171,3; 169,8; 166,7; 136,0; 129,3; 128,4; 126,9; 69,2; 60,3; 53,7; 53,3; 37,8; 22,9; 21,7; 21,6; 14,1; [α]25 D = -1,73° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-fenilalaninato de n-butilo (8, C18H25NO5)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,48 (d; J = 8,2 Hz; 2H); 7,31-7,09 (m; 5H); 6,19 (t; J = 7,7 Hz; 1H); 4,91-4,77 (m; 5H); 4,09 (dtd; J = 12,5; 7,2; 6,5; 3,6 Hz; 7H); 3,13 (d; J = 7,5 Hz; 2H); 1,97 (s; 3H); 1,61-1,50 (m; 7H); 1,36-1,22 (m; 9H); 1,22-1,07 (m; 1H); 0,91 (q; J = 7,0 Hz; 3H); RMN 13C (100 MHz; CDCl3) δC 171,9; 171,4; 167,0; 166,7; 136,0; 129,3; 129,3; 128,5; 127,1; 77,5; 77,2; 76,8; 65,4; 53,7; 53,2; 37,9; 30,5; 23,1; 19,1; 13,7; [α]25 D = +8,86° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-tirosinato de metilo (9, C15H19NO6)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,58 (d; J = 7,8 Hz; 1H); 7,26 (s; 1H); 6,97 (d; J = 8,4 Hz; 2H); 6,72 (d; J = 8,1 Hz; 2H); 4,84 (td; J = 6,9; 5,5 Hz; 1H); 4,17 (q; J = 7,2 Hz; 2H); 3,73 (s; 3H); 3,29 (d; J = 3,4 Hz; 2H); 3,13-2,95 (m; 2H); 1,25 (t; J = 7,2 Hz; 3H); RMN 13C (100 MHz; CDCl3) δC 172,0; 169,0; 165,3; 155,5; 130,4; 127,2; 115,7; 61,9; 53,8; 52,6; 41,1; 37,2; 14,1; [α]25 D = +6,89° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-tirosinato de etilo (10, C16H21NO6)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 7,51 (d; J = 7,8 Hz; 1H); 7,04-6,96 (m; 2H); 6,76-6,68 (m; 2H); 5,57 (s; 1H); 4,82 (dt; J = 7,8; 6,1 Hz; 1H); 4,18 (qd; J = 7,1; 1,4 Hz; 5H); 3,29 (d; J = 2,4 Hz; 2H); 3,132,97 (m; 2H); 1,28-1,24 (m; 6H); RMN 13C (100 MHz; CDCl3) δC 171,8; 169,3; 165,3; 155,4; 130,9; 128,1; 115,9; 62,2; 54,1; 41,6; 37,6; 14,6;[α]25 D =+11,25° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-tirosinato de isopropilo (11, C17H23NO6)
Aceite amarillo; RMN 1H (400 MHz; CDCl3) δH 7,60 (d; J = 7,7 Hz; 1H); 6,95 (d; J = 8,2 Hz; 2H); 6,70 (d; J = 8,7 Hz; 2H); 4,98 (p; J = 6,4 Hz; 7H); 4,74 (q; J = 6,5 Hz; 1H); 4,12 (q; J = 7,1 Hz; 2H); 3,27 (d; J = 2,5 Hz; 2H); 3,08-2,93 (m; 2H); 1,20 (t; J = 6,4 Hz; 9H); RMN 13C (100 MHz; CDCl3) δC 171,0; 168,8; 165,4; 155,7; 130,4; 126,8; 115,5; 77,5; 77,2; 76,8; 69,6; 61,8; 53,9; 41,2; 37,1; 21,8; 14,0; [α]25 D = +4,39° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-tirosinato de n-butilo (12, C18H25NO6)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,83 (s; 1H); 7,63 (d; J = 7,6 Hz; 1H); 6,91 (d; J = 8,3 Hz; 2H); 6,69 (d; J = 8,1 Hz; 2H); 4,76 (q; J = 6,5 Hz; 1H); 4,14-4,01 (m; 5H); 3,25 (d; J = 2,5 Hz; 2H); 3,072,91 (m; 2H); 1,60-1,47 (m; 4H); 1,33-1,24 (m; 3H); 1,18 (t; J = 7,2 Hz; 2H); 0,86 (t; J = 7,4 Hz; 3H); RMN 13C (100 MHz; CDCl3) δC 171,5; 168,7; 165,6; 155,7; 130,2; 126,6; 115,5; 77,5; 77,2; 76,8; 65,5; 61,7; 53,8; 41,1; 36,9; 30,3; 18,9; 13,9; 13,6; = +6,70° ± 0,01 (c 0,6; MeOH). ESI-HRMS m/z [M+Na]+, 374,1574 (calcd. 374.1574).
N a -(3-etoxi-3-oxopropanoil)-L-alaninato de metilo (13, C9H15NO5)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,64 (d; 1H); 4,58 (q; J = 7,2 Hz; 1H); 4,19 (q; J = 7,2 Hz; 2H); 3,73 (s; 3H); 3,32 (s; 2H); 1,41 (d; J = 7,2 Hz; 3H); 1,27 (t; J = 7,2 Hz; 3H); RMN 13C (100 MHz; CDCl3) δC 173,2; 169,3; 164,8; 61,8; 52,6; 48,3; 41,1; 18,3; 14,1; [α]25 D = -17,97° ± 0,01 (c 0,6; MeOH).
N a -(3-etoxi-3-oxopropanoil)-L-alaninato de etilo (14; C10H17NO5)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,64 (d; J = 7,6 Hz; 1H); 4,57 (q; J = 7,2 Hz; 1H); 4,20 (qd; J = 7,2; 3,2 Hz; 3H); 3,32 (s; 2H); 1,43 (d; J = 7,2 Hz; 3H); 1,28 (td; J = 7,1; 4,7 Hz; 6H); RMN 13C (100 MHz; CDCl3) δC 172,8; 169,3; 164,7; 61,8; 61,7; 48,4; 41,2; 18,4; 14,2; 14,2, [α]25 D = -21,15° ± 0,01 (c 0,6; MeOH). ESI-HRMS m/z [M+Na]+; 254,1007 (calcd, 254,0999).
N a -(3-etoxi-3-oxopropanoil)-L-alaninato de isopropilo (15, C11H19NO5)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,71 (dd; J = 16,4; 7,2 Hz; 1H); 4,96 (q; J = 6,2 Hz; 1H); 4,51-4,37 (m; 1H); 4,05 (q; J= 7,1 Hz; 2H); 3,23 (d; J = 3,0 Hz; 1H); 1,32 (dd; J = 9,7; 7,2 Hz; 4H); 1,18 (dd; J= 8,5; 5,4 Hz; 6H); RMN 13C (100 MHz; CDCl3) δC 172,3; 169,9; 167,0; 77,5; 77,2; 76,8; 69,1; 48,5; 42,5; 21,7; 17,8; [α]25 D = -20,98° ± 0,01 (c 0,6; MeOH). ESI-HRMS m/z [M+Na]+, 268,1161 (calcd. 268,1155).
N a -(3-etoxi-3-oxopropanoil)-L-alaninato de n-butilo (16, C12H21NO5)
Aceite amarillo pálido; RMN 1H (400 MHz; CDCl3) δH 7,78 (d; J = 7,2 Hz; 1H); 6,51 (d; J = 7,5 Hz; 1H); 4,48 (dt; J = 14,6; 7,3 Hz; 2H); 4,06 (td; J = 6,6; 3,7 Hz; 4H); 3,23 (s; 1H); 1,60-1,51 (m; 4H); 1,36-1,26 (m; 8H); 0,86 (t; J = 7,4; 2,0 Hz; 3H); RMN 13C (100 MHz; CDCl3) δC 173,4; 172,9; 169,9; 167,1; 65,3; 48,3; 48,1; 42,4; 30,5; 23,0; 19,0; 17,7; 13,6; [α]25 D = -19,90° ± 0,01 (c 0,6; MeOH). ESI-HRMS m/z [M+Na]+, 282,1322 (calcd. 282.1312).
Actividad antifúngica a partir de la inhibición del crecimiento micelial de Fusarium oxysporum
La evaluación de la actividad antifúngica de los compuestos 1-16 se realizó por medio de la medición del crecimiento radial del micelio del fitopatógeno Fusarium oxysporum siguiendo una metodología previamente reportada [35]. Brevemente, en cada pozo de una placa de vidrio de 12 pozos se adicionó agar de papa-dextrosa (PDA) suplementado separadamente con los compuestos 1-16 y controles a evaluar, utilizando concentraciones en un rango de 0,1-100 mM, en un arreglo por triplicado para cada compuesto y concentración. Una vez adicionado el agar suplementado en cada pozo, se inoculó con un fragmento cilíndrico de 3 mm de diámetro y 1 mm de altura, proveniente de un cultivo activo de F. oxysporum. El hongo se dejó crecer en cámara húmeda hasta que el micelio del control absoluto (i. e., sin tratamiento) llegó hasta el borde del pozo. Luego de la incubación, se registraron las áreas de crecimiento micelial mediante el software ImageJ y se calcularon los porcentajes de inhibición mediante la siguiente fórmula: % de inhibición = (CA-T)*(100/CA), donde CA representa el diámetro del crecimiento micelial para el control absoluto y T representa el diámetro del crecimiento micelial del tratamiento respectivo. Los datos de inhibición (%) se emplearon para construir las correspondientes curvas dosis-respuesta con el fin de calcular la concentración inhibitoria media (IC50, expresada en mM) para cada compuesto, usando una regresión no lineal en el programa GraphPad Prism versión 9.0 (GraphPad Software, San Diego, CA, EE. UU.) para Windows.
Resultados y discusión
Para la síntesis de amidas N-sustituidas se planteó un modelo de estudio buscando aplicar la metodología optimizada para la síntesis de enaminas previamente descrita en la literatura [8]. En esta etapa inicial, los 2-aminoésteres fueron obtenidos mediante la reacción entre el z-triptófano y el respectivo alcohol (MeOH, EtOH) en cloruro de trimetilsililo (TMSCl), para posteriormente adicionar trietilamina (Et3N) y malonato de dietilo (DEM). La mezcla de reacción se calentó bajo reflujo hasta que se evidenció consumo mayoritario de los reactivos (tiempo de reacción entre 20 y 30 h). El perfil cromatográfico por CCD evidenció la presencia de múltiples bandas difíciles de separar y una banda en el origen correspondiente al aminoácido de partida. Además, el DEM fue recuperado con acetato de etilo a partir del crudo de reacción. Estos resultados permitieron concluir que el DEM es poco reactivo bajo estas condiciones y que, posiblemente, los 2-aminoésteres de alquilo derivados del L-triptófano se hidrolizaron al permanecer en una atmósfera húmeda y en tiempos prolongados de reacción, lo que permitió deducir que la metodología evaluada no era sintéticamente adecuada. Por tal motivo, fue necesario explorar otros métodos de síntesis alternativos orientados a la activación del DEM.
El uso de ácidos de Lewis se emplea para activar el grupo carbonilo de los ésteres por polarización, ya que es uno de los métodos más confiables para inducir la formación de amidas a partir de ésteres [36]. Por otro lado, la activación de esta clase de compuestos puede realizarse con un nucleófilo tipo amina; por ejemplo, DMAP. Este compuesto ha sido utilizado ampliamente para muchas reacciones orgánicas, como la reacción de Baylis-Hillman, la síntesis de β-lactamas monocíclicas de N-sulfonilo [37] y de β-cetoamidas a partir de β-cetoésteres [38]. Teniendo en cuenta estos antecedentes, después de obtener los 2-aminoésteres de alquilo bajo irradiación por MW, se evaluó el uso tanto de AlCl3 como de DMAP, para lo cual se plantearon dos experimentos: en el primero, se preparó una mezcla con DEM, tolueno, AlCl3 y ácido acético (método A), la cual se dispuso en un tubo de reacción y se irradió bajo MW a diferentes temperaturas (80, 100, 120 y 150 °C) y a diferentes tiempos de reacción. Los resultados del seguimiento de las reacciones por CCD permitieron establecer que el DEM reaccionó completamente a 150 °C durante 20 min. Después de este tiempo, la reacción se acidificó con HCl diluido, hasta pH entre 5-6, para liberar el AlCl3 y, posteriormente, se realizó extracción con acetato de etilo (5x 10 mL). El crudo de reacción se concentró a sequedad y fue purificado por cromatografía en columna, separando el producto mayoritario; en el segundo experimento (método B), se empleó DMAP como auxiliar nucleofílico. Así, después de obtener los 2-aminoésteres de alquilo, se adicionó Et3N para neutralizar el exceso de HCl y liberar el grupo amino y mantenerlo como base libre, y garantizando medio básico. Posteriormente, se adicionó DEM y DMAP y se irradió bajo MW a distintas temperaturas (100, 120 y 150 °C) y a diferentes tiempos de reacción. Los resultados del seguimiento de las reacciones por CCD indicaron que el DEM reaccionó completamente a 140 °C y 20 min, y se formó el mismo producto que en el método A.
A continuación, se describe la caracterización por espectroscopía del compuesto 1, el cual fue aislado y purificado tanto para el método A como para el método B. En el espectro de RMN 1H del compuesto 1 se observaron señales que corresponden a hidrógenos aromáticos del grupo indol en la región de desplazamiento químico entre 7,00 y 7,50 ppm. A δH 4,93 ppm aparece una señal como multiplete que corresponde al hidrógeno del carbono quiral; la señal a campo alto como singlete a δH 3,63 ppm fue asignada al grupo metoxilo del éster. El fragmento etoxilo del malonato de dietilo puede verse como señales a δH 4,12 ppm (grupo CH2O) y la señal triplete a δH 1,22 ppm del grupo CH3. La señal tipo doblete a δH 7,50 ppm fue asignada al protón del grupo NH de la amida. La señal a δH 3,28 ppm, que aparece doblete, corresponde al CH2 adyacente a los grupos C=O (Figura 1a). En el espectro de RMN 13C del compuesto 1 se pueden apreciar 18 señales, las cuales se asignan a continuación. Las señales a δC 172,1; 168,9 y 164,2 ppm corresponden a los grupos carbonilo, mientras que las señales a δC 136,2; 127,6; 122,9; 122,3; 119,7; 118,7; 111,3 y 110,1 ppm fueron asignadas a los carbonos aromáticos del grupo indol. Así mismo, se observaron dos señales para el grupo amida, i. e., el átomo de carbono directamente unido al grupo NH que resuena a δC 163,0 ppm y el grupo CH2 adyacente a los dos C=O que resuena a δC 41.4 ppm (Figura 1b).
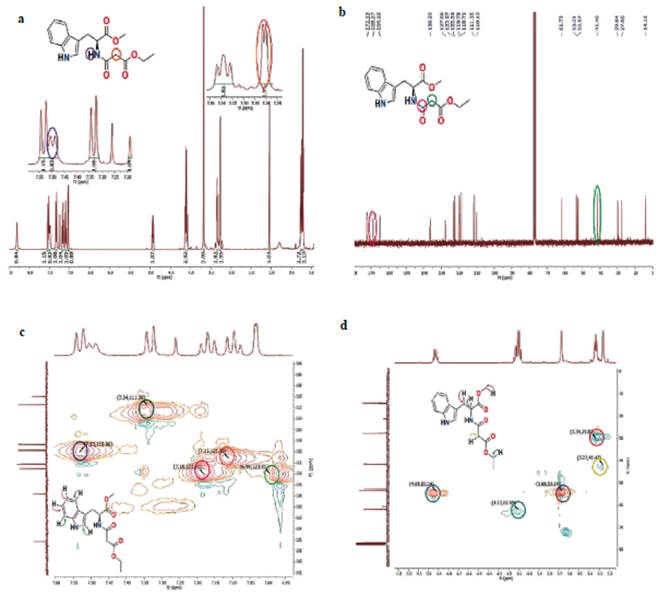
Figura 1 Experimentos RMN uni y bidimensionales del compuesto 1, a) RMN 1H, b) RMN 13C, c) HSQC, d) HMBC.
El experimento HSQC evidenció que la señal a δC 7,50 ppm no muestra ninguna correlación, lo que indica que esta señal corresponde al NH de la amida. La señal a δC 3,27 ppm mostró una correlación directa con la señal a δC 41,4 ppm, lo cual confirmó la asignación realizada al grupo CH2 adyacente a los grupos C=O (Figura 1c). Una vez establecidas las condiciones óptimas de reacción, estas fueron empleadas en las reacciones con los restantes precursores. Los rendimientos obtenidos para el método A se presentan en color azul y para el método B en color rojo (Figura 2). Los compuestos 2-16 fueron análogamente elucidados. Sus datos espectroscópicos se reportan en la sección Materiales y métodos. Los espectros de masas de alta resolución (HRMS) de los compuestos 12, 14, 15 y 16, los cuales fueron seleccionados aleatoriamente del grupo total de compuestos para esta determinación, mostraron señales para los iones [M+Na]+ con errores menores que 5 ppm, lo cual confirmó la estructura propuesta para los compuestos 1-16.
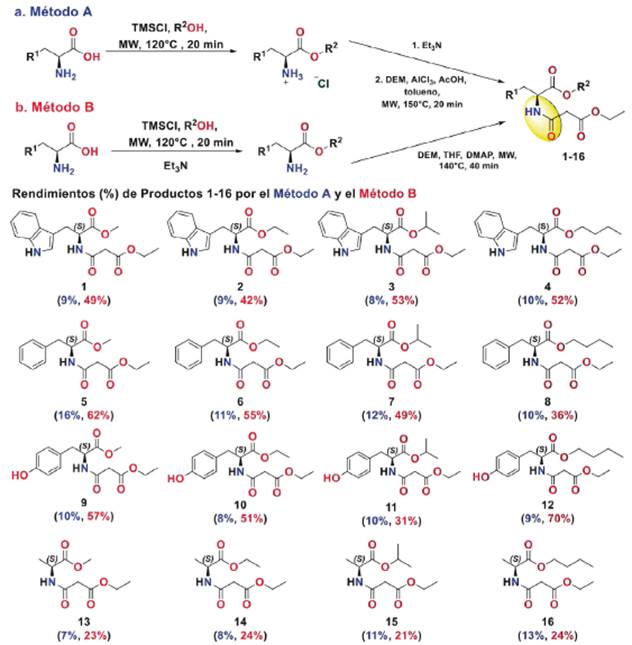
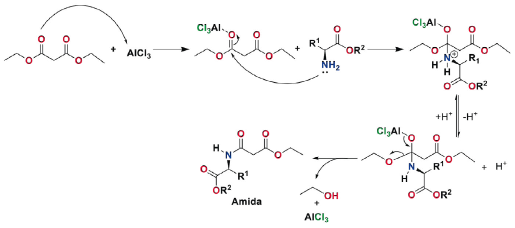
Los rendimientos de los compuestos 1-16 variaron con respecto a la metodología empleada, a pesar de que se empleó la irradiación por MW. En el método A se observa que los rendimientos son menores del 15%, mientras que para el método B se evidencia una mejora significativa (2060%). Los bajos rendimientos obtenidos por el método A pueden explicarse debido a la posible formación de un quelato entre el DEM y el centro metálico, lo que forma un complejo estable [39]. Es sabido que un centro metálico que se comporte como ácido de Lewis, como el AlCl3, tiene una fuerte tendencia a los alcóxidos metálicos para formar oligómeros con "puente alcóxido" para alcanzar números de coordinación más altos. El grado de oligomerización está inversamente correlacionado con el tamaño del grupo alcóxido. Esta tendencia a formar oligómeros se mantiene después de la modificación con otros ligandos orgánicos como compuestos dicarbonílicos como acetilacetona, acetoacetato de etilo incluso, DEM. Lichtenberger y colaboradores [40] han reportado que, aunque DEM y otros ésteres de alquilo, derivados del ácido malónico, tienen una menor reactividad hacia los alcóxidos de aluminio a temperatura ambiente en comparación con los 2-cetoésteres, a temperaturas más altas reaccionan formando complejos estables. Dichos compuestos pueden formarse al reaccionar Al(OR)3 (adicionado como reactivo o formado in situ) con DEM en una relación estequiométrica 1:3. Dado que en nuestras condiciones de reacción estudiadas se cuenta con presencia de grupos RO- y de un exceso de DEM, es de esperar la formación de dichos complejos, lo cual reduce significativamente el porcentaje de rendimiento de los compuestos 1-16. Por lo tanto, a partir de los resultados por este método se deduce que la eficiencia de la reacción no está dependiendo de la naturaleza estructural del aminoácido, dado que los rendimientos son similares al comparar cada uno de dichos precursores. El mecanismo de reacción se presenta en la Figura 3. La activación catalizada por el AlCl3 del grupo carboxilato del malonato de dietilo (DEM) inicia la reacción tras la formación del quelato, lo cual hace que se incremente la electrofilicidad del grupo C=O coordinado con el aluminio y, de esta manera, permite el ataque nucleofílico del grupo NH2 del 2-aminoéster de alquilo. Una vez ocurre este ataque, se da la formación de un intermediario catiónico, el cual cuenta con un hidrógeno ácido sobre el grupo NH2 + que posteriormente reacciona con Et3N, lo que da lugar a la formación del intermediario tetraédrico. Sin embargo, este intermediario no es estable y cuenta con un buen grupo saliente como el alcóxido, el cual se libera y conlleva la formación de la amida.
Para el método B, el hecho de emplear un auxiliar nucleofílico, como la DMAP, fue esencial para mejorar los rendimientos ya que, en ausencia de esta, la reacción no procedió; esto demostró una participación nucleofílica de DMAP en el avance de la reacción. Este reactivo ha sido empleado en diversas reacciones, de las cuales sobresalen las de esterificación de Steglich y la de Yamaguchi [41]-[43]. En la primera, se lleva a cabo la conversión de sustratos estéticamente impedidos usando DCC (diciclohexilcarbodiimida) y el ácido carboxílico puede formar un intermedio de O-acilisourea, que ofrece una reactividad similar al anhídrido de ácido carboxílico correspondiente. El alcohol puede agregarse al ácido carboxílico activado para formar la diciclohexilurea (DHU) estable y el éster. Sin embargo, los nucleófilos fuertes, por ejemplo las aminas, reaccionan fácilmente con la O-acilisourea, lo cual se explica por el comportamiento nucleofílico del DMAP, más fuerte que el alcohol, que reacciona con la O-acilisourea dando lugar a una amida reactiva. Este intermediario tipo amida no puede formar productos secundarios intramoleculares, pero reacciona rápidamente con los nucleófilos del medio, en este caso particular, con los alcoholes. Así, DMAP actúa como un reactivo de transferencia de acilo al contar con un grupo acilo polarizado. Como el DMAP es un buen grupo saliente, permite una reacción rápida con el nucleófilo del medio. En las reacciones estudiadas en el presente estudio, DEM y DAMP reaccionan dando lugar a la formación de un intermediario similar, el cual es más reactivo que el grupo éster del DEM, permitiendo el posterior ataque nucleofílico de la amina del éster de alquilo derivado del aminoácido y la salida del DMAP para dar la formación de la amida iV-alquilsustituida. Para esta metodología se observó que, empleando el aminoácido z-alanina como precursor, los porcentajes de rendimiento fueron los más bajos (< 25%), lo cual puede ser explicado a partir de reacciones colaterales, tipo condensación de Claisen cruzada, que pueden ocurrir por desprotonación del hidrógeno sobre el carbono quiral, por medio de una base orgánica. Esto no se evidenció en los restantes aminoácidos; se entiende que dicho átomo de hidrógeno se encuentra menos expuesto al ataque de la base cuando la cadena lateral del aminoácido es un grupo más voluminoso que el grupo metilo. El mecanismo de reacción propuesto para el método B se presenta en la Figura 4. El DMAP hace un ataque nucleofílico sobre uno de los carboxilatos del DEM, formando un intermediario tetraédrico zwitteriónico que libera un grupo alcóxido para generar un catión 4-(dimetilamino)-1-(3-etoxi-3-oxopropanoil)piridinio estabilizado por resonancia. Posteriormente, se lleva a cabo el ataque nucleofílico del 2-aminoéster de alquilo sobre el grupo C=O tipo amida, formando el intermediario hemiacetalaminal. Finalmente, dadas las condiciones básicas del medio, se da la salida del fragmento DMAP y, por ende, la formación de la amida.
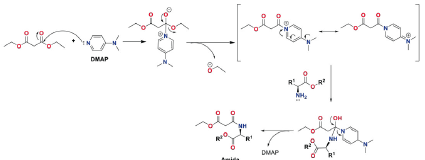
Figura 4 Mecanismo de reacción propuesto para la síntesis de amidas yV-alquilsustituidas (método B).
La evaluación de la actividad antifúngica in vitro sobre F. oxysporum se realizó empleando el ensayo de medio suplementado a microescala [35]. Los resultados se expresaron como la concentración inhibitoria media (IC50 en mM) para cada compuesto, utilizando una regresión no lineal, cuyos valores se presentan en la Tabla 1. Los valores de IC50 medidos para las amidas iV-alquilsustituidas 1-16 oscilaron entre 0,40 y 29,1 mM.
Tabla 1 Valores de IC50 (mM) para las amidas TV-alquilsustituidas 1-16 sobre F. oxysporum.
| Compuesto | IC50 (IC)a |
|---|---|
| 1 | 3,43 (2,58-4,52) |
| 2 | 0,40 (0,30-0,50) |
| 3 | 3,02 (2,28-3,98) |
| 4 | 2,28 (1,67-3,06) |
| 5 | 8,80 (5,82-13,99) |
| 6 | 16,9 (12,3-25,3) |
| 7 | 2,55 (2,15-3,02) |
| 8 | 16,9 (11,6-21,6) |
| 9 | 19,1 (14,8-25,9) |
| 10 | 0,44 (0,34-0,55) |
| 11 | 3,76 (2,48-5,71) |
| 12 | 5,46 (3,61-8,52) |
| 13 | 8,91 (6,05-13,6) |
| 14 | 17,2 (12,7-24,8) |
| 15 | 29,1 (21,1-43,8) |
| 16 | 25,4 (16,6-46,1) |
| pb | 0,02 (0,01-0,03) |
a Los valores de IC50 están expresados en mM, con su intervalo de confianza (CI) al 95% entre paréntesis; b p = prochloraz como control positivo.
Los resultados de la actividad antifúngica in vitro de las amidas N-alquilsustituidas mostraron que 2 y 10 presentaron menores valores de IC50 (0,40 y 0,44 mM, respectivamente). Estos resultados permitieron establecer que, en algunos casos, el tamaño del fragmento aromático del aminoácido podría mejorar la actividad frente a F. oxysporum. Sin embargo, la actividad antifúngica se vio influenciada por la cadena lateral del aminoácido precursor, dado que los compuestos más activos presentan tanto grupo 1H-indol como 4-hidroxifenilo. Esto sugiere que la presencia de un sistema aromático y un grupo, tanto donor como aceptor de puente de hidrógeno (i. e., NH u OH), favorece la actividad antifúngica, debido a la interacción plausible con algunos objetivos bioquímicos relevantes en el metabolismo de Fusarium oxysporum, como se ha reportado previamente [8]. Estos resultados también pueden explicarse a favor de la presencia del grupo amida acíclica como un bioisóstero no clásico de ciertos heterocíclicos (e. g., triazol, tiazol, imidazol) y ditiocarbamatos, comúnmente presentes en fitoalexinas de crucíferas [44], el cual puede modular la actividad biológica como sustitutos debido al favorecimiento en el tamaño molecular, forma, distribución electrónica, polarizabilidad, lipofilicidad, polaridad, entre otros [45]. En este sentido, diversas investigaciones han demostrado que compuestos que contienen grupo amida han sido evaluados contra fitopatógenos fúngicos [46]. Wu y colaboradores [47] sintetizaron diferentes compuestos derivados de amida de pirazol que tienen un fragmento de hidrazona. Estos compuestos se evaluaron frente a F. oxysporum; sus porcentajes de inhibición fueron de alrededor del 40% [47]-[49]. Recientemente, nuevos derivados de amidas de pirazol fueron sintetizados por Zhang y colaboradores, los cuales fueron evaluados a escala in vitro sobre F. oxysporum a una concentración de 20 mg/mL. Los resultados obtenidos mostraron que este tipo de compuestos presentaron una moderada actividad antifúngica con porcentajes de inhibición entre 20 y 60% [50]. Pese a que resulta necesario involucrar un espacio químico más amplio para establecer un modelo de correlación estructura-actividad que permita establecer con mayor fidelidad el efecto del grupo amida, los resultados discutidos en este manuscrito evidencian un comportamiento promisorio de este grupo funcional en derivados de 2-aminoácidos hacia un rol como toxóforo contra F. oxysporum en razón de su bioisosterismo no clásico en fitoalexinas.
Conclusiones
En el presente estudio se obtuvieron nuevas amidas N-sustituidas 1-16 empleando dos metodologías de síntesis. El uso de DMAP generó mejores porcentajes de rendimiento respecto al uso de AlCl3 como ácido de Lewis. Los resultados de la actividad antifúngica frente a F oxysporum de las amidas N-sustituidas 1-16 permitieron establecer que la variabilidad estructural influencia la actividad, principalmente debido a la presencia de grupos 1H-indol y 4-hidroxifenilo en los ésteres de etilo de los 2-aminoácidos respectivos (i. e., compuestos 2 y 10). Lo anterior demostró el potencial de esta clase de derivados de 2-aminoácidos convencionales como bioactivos antifúngicos contra F. oxysporum debido al bioisosterismo no clásico de la amida acíclica con fragmentos heterocíclicos de fitoalexinas.

