










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombian Journal of Anestesiology
Print version ISSN 0120-3347
Rev. colomb. anestesiol. vol.46 no.1 Bogotá Jan./Mar. 2018
https://doi.org/10.1097/cj9.0000000000000013
REPORTE DE CASO
Manejo anestésico en paciente pediátrico con distroña miotónica tipo 1. Reporte de caso
a Departamento de Anestesiología y Reanimación, Hospital General Universitario Gregorio Marañón, Madrid, España
b Departamento de Anestesiología y Reanimación, Hospital Clínico San Carlos. Madrid, España
La distrofia miotónica es una enfermedad de las fibras musculares que cursa con pérdida de masa muscular y cuya característica principal es la miotonía, que describe la relajación muscular lenta tras una contracción muscular, situación agravada por estrés, dolor, frío, o por la administración de succinilcolina. Como toda enfermedad muscular, es considerada multisistémica, con afectación cardíaca y respiratoria en la mayoría de los casos, lo cual deberá tenerse en cuenta a la hora de elaborar un plan anestésico. Además, se debe considerar la posible relación con el desarrollo de hipertermia maligna o rabdomiólisis asociada a algunas enfermedades musculares. El caso que presentamos es un ejemplo del manejo anestésico de estos pacientes evitando los posibles desencadenantes de una crisis miotónica.
Palabras clave: Distrofia Miotónica; Músculo Esquelético; Niño; Anestesia; Hipotonia Muscular
Myotonic dystrophy is a disease affecting the muscle fibers with loss of muscle mass. The principal characteristic of the disease is myotony or slow muscle relaxation following muscle contraction that is further aggravated as a result of stress, pain, cold, or by the administration of succinylcholine. Similar to other muscle pathologies, myotonic dystrophy is considered a multisystem disorder, usually with cardiac and respiratory involvement, a fact to be kept in mind when planning anesthesia. Moreover, there is a potential association with malignant hyperthermia or rhabdomyolysis associated with some muscle diseases. The case herein discussed is an example of the management of anesthesia in this group of patients to avoid the potential triggers of a myotonic crisis.
Keywords: Myotonic Dystrophy; Muscle; Skeletal; Child; Anesthesia; Muscle Hypotonia
Introducción
La distrofia miotónica tipo 1 (DM1), clásicamente conocida como enfermedad de Steinert, es una enfermedad multi-sistémica que afecta predominantemente al musculo esquelético, sistema nervioso central, respiratorio, gastrointestinal, cardíaco y endocrino en neonatos, niños y adultos. La DM1 tiene una herencia autosómica dominante con una penetrancia variable y se ocasiona por la expansión repetida del triplete CTG de un segmento de ADN no codificante del gen DMPK en el cromosoma 19q13.3. La incidencia y la prevalencia de la DM1 son aproximadamente 13/10.000 y 2-14/100.000 respectivamente. Estos pacientes presentan un mayor riesgo anestésico debido principalmente a la afectación muscular, que empeora la función respiratoria, y a la clínica multiorgánica.1,2
Caso clínico
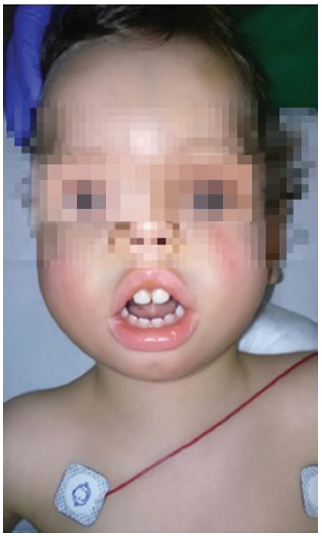
Presentamos un paciente varón de 22 meses de edad y 9 kg de peso que es intervenido de forma programada de orquidopexia bilateral. Como antecedentes destacan polihidramnios en el embarazo y nacimiento por cesárea por riesgo de pérdida de bienestar fetal en la semana 41+3. El Apgar fue 1/10 asociado a aspiración meconial que requirió intubación orotraqueal y vigilancia en UCI postnatal. Al nacimiento el paciente fue diagnosticado de DM1, así como la madre, que no presentaba evidencia de fenómeno miotónico. El paciente presenta facies inexpresiva, boca de pez, retrognatia y frente prominente, típica de DM 1 (Figura 1), hipotonía, ectasia piélica bilateral, pies equinos reducibles y episodios de extra-sistolia auricular con conducción aberrante. Su tratamiento habitual consiste en budesonida, montelukast, flecainida y atenolol.
En quirófano se monitoriza tensión arterial no invasiva, electrocardiograma, pulsioximetría y temperatura esofágica. Se realiza inducción anestésica inhalatoria con sevofluorano y colocación de mascarilla laríngea #1.5, iniciándose ventilación asistida con presión de soporte y monitorización de CO2 espirado, manteniéndose este agente halogenado con una concentración alveolar mínima de 2.8% durante el procedimiento. Para el control del dolor, se realiza una punción caudal y administración en bolo (9ml) de bupivacaína 0.125% y lidocaína 0,5%, con colocación ecoguiada de catéter hasta nivel T12 y perfusión posterior de ropivacaína 0.1%. Durante la intervención se administra remifentanilo en perfusión continua a 0.1mcg/kg/min. La intervención cursa sin incidencias, despertando al paciente en quirófano. Posteriormente es llevado a la UCI pediátrica para vigilancia durante las primeras 24 horas, manteniendo perfusión continua de ropivacaína 0.1% por catéter caudal, para control del dolor postoperatorio, sin que se produzca ninguna complicación.
Discusión
En el manejo anestésico de estos pacientes es importante tener en cuenta una serie de consideraciones especiales, además de evitar todos los posibles desencadenantes de una crisis miotónica.
La premedicación con benzodiazinas está contraindicada en estos pacientes por su afección muscular y respiratoria, presentando un elevado riesgo de depresión respiratoria, por lo que el paciente llega a quirófano sin premedicación.1,2
Los anestésicos halogenados pueden ser utilizados como inductores y anestésicos de mantenimiento de forma segura ya que, a pesar de encontrarse su gen codificante en el mismo cromosoma que la hipertermia maligna, no se ha relacionado el uso de este tipo de anestésicos con el desarrollo de este grave cuadro3) en estos pacientes. Además, en presencia de una crisis miotónica, los halogenados contribuyen a mitigar la crisis, mientras que los relajantes no despolarizantes no son de utilidad, puesto que el origen de la contracción se debe a hiperexcitabilidad de la membrana.
El uso de opioides para el control del dolor puede agravar problemas respiratorios subyacentes, siendo el remifentanilo una buena opción para el control del dolor agudo intraoperatorio por su vida media corta. Respecto a la anestesia regional ha sido descrita con éxito en la literatura para disminuir la administración de opiodes en el manejo del dolor intraoperatorio y postoperatorio.1-3
Debe evitarse la hipotermia, que podría desencadenar una crisis miotónica, manteniendo una adecuada temperatura ambiental, además de colocación de manta térmica y calentador de sueros si fuera necesario.1,3
La succinilcolina está contraindicada por la capacidad de desencadenar miotonías dependientes de la dosis, y el uso de relajantes musculares no despolarizantes de acción corta está permitido ajustando la dosis y monitorizando su efecto.1,2
En cuanto a la distrofia miotónica tipo 2 o miopatía miotónica proximal, las consideraciones en el manejo anestésico serían similares, si bien se trata de pacientes que pueden mantenerse asintomáticos hasta la edad adulta y presentan un menor riesgo anestésico.2
Para concluir, observar que el manejo de cada paciente debe ser personalizado, sirviendo este caso para enfatizar en la importancia en estos pacientes de una exhaustiva valoración preanestésica, un cuidadoso manejo intraoperatorio, evitando la hipotermia, estrés y dolor y estando permitidos los anestésico volátiles, relajantes musculares no despolarizantes, opiodes de vida media corta y anestesia locorregional, además de una vigilancia intensiva postoperatoria.
Responsabilidades éticas
Protección de personas y animals. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
REFERENCIAS
1. Ho G, Cardamone M, Farrar M. Congenital and childhood myotonic dystrophy: current aspects of disease and future directions. World J Clin Pediatr 2015;4:66-80. [ Links ]
2. Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Pediatr Anesth 2013;23:794-803. [ Links ]
3. Pérez Ferrer A, Calvo Vecino JM. El niño con patologías muscu-loesqueléticas. En: Manual de Anestesiología pediátrica. Bogotá: Editorial Médica Panamericana; 2016. [ Links ]
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
