











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink
En Colombia, según análisis epidemiológicos, para el periodo comprendido entre 2007 y 2011, los tumores primarios del sistema nervioso central (SNC) presentaron una incidencia anual ajustada por edad de 3,4 casos por cada 100.000 habitantes hombres y de 2,5 casos por cada 100.000 mujeres, con una mortalidad de 2,5 y 1,9 por cada 100.000 hombres y mujeres, respectivamente (1). Ello resulta preocupante, ya que refleja bajas tasas de supervivencia, las cuales se han estimado como una sobrevida a 3 años del 15,2 % en población pediátrica (2).
Un grupo infrecuente y heterogéneo de neoplasias primarias del SNC son los tumores de células germinales intracraneales (TCGI). Representan entre el 1 % y el 3 % de los tumores malignos neurológicos; ocurren con mayor frecuencia en pacientes pediátricos, masculinos y asiáticos (3). El origen de las células tumorales es controvertido. Lo más aceptado es que provienen de células primordiales que, durante su migración al esbozo gonadal, pueden establecerse ectópicamente en el SNC y experimentar una transformación neoplásica (4). Aproximadamente, un 65 % de tales tumores se clasifica como germinoma, el tercio restante hace parte de un conjunto amplio y diverso de tumores, denominados no germinomas (5). La clasificación resulta elemental para el manejo, ya que guía el plan diagnóstico y terapéutico; razón por la que esta revisión se presenta subdividida según tal clasificación.
La rareza de los TCGI ha limitado nuestro conocimiento sobre ellos, así como entorpeciendo el avance diagnóstico y terapéutico. Además, es poca (casi nula) la divulgación científica que se ha hecho sobre el tema en los países de occidente, como Colombia. Dicha problemática es la principal motivación para este trabajo, mediante el cual se busca exponer de forma sucinta los principales aspectos clínico-patológicos de los TCGI, en procura de contribuir a su mejor comprensión.
Metodología
Se efectuó una búsqueda sistematizada de información mediante la estrategia PICO. Identificado el problema, se estructuró la pregunta: ¿cuáles son los principales tumores germinales del SNC, sus características básicas y abordaje diagnóstico-terapéutico? Las bases de datos electrónicas utilizadas fueron Pubmed, ScienceDirect, Cochrane y Google Scholar. Se seleccionaron artículos originales, revisiones, casos clínicos, libros y guías de práctica clínica, escritas en inglés o español, en la medida de lo posible, publicadas en los últimos años.
Tumores de células germinales intracraneales
Los TCGI afectan principalmente a niños, adolescentes y adultos jóvenes, con una mediana de 10 años. Casi el 90 % de los casos se presentan antes de los 20 años de edad (3,6). La relación entre afectados hombres versus mujeres es 3:1 (7).
Este grupo hace parte de varios tipos de neoplasias poco estudiadas y, en cierto modo, subvaloradas en el mundo occidental, no así en el continente asiático, donde se cree que tienen mayor prevalencia. Algunos estudios han demostrado incidencias comparativas del 14,3 % en Japón, del 14,0 % en Taiwán, del 11,2 % en Corea, contra un 2,3 % en Estados Unidos (8,9,10,11). Otros estudios hallaron incidencias similares entre los países asiáticos y Estados Unidos (7,12,13). Finalmente, es confuso definir el patrón de incidencia de los TGCI en el mundo; lo cierto es que la comunidad científica asiática es la más comprometida con la actividad académica sobre el tema.
La variedad de TCGI ha sido clasificada por la Organización Mundial de la Salud. En su última modificación de 2016, los divide en germinomas y tumores de células germinales no germinomatosas (NGGCT). A los últimos pertenecen el teratoma (maduro, inmaduro y teratoma con transformación maligna), el carcinoma embrionario, el tumor del saco vitelino, el coriocarcinoma y los TGC mixtos (14).
Las células germinales primordiales, durante el periodo embrionario temprano, pasan de un estado pluripotente a un estado diferenciado, y así quedan destinadas a convertirse en gametos maduros. En este periodo se ubican en el endodermo, de donde migran hasta las gónadas indiferenciadas, y ahí puede volverse malignos, por lo que los TCG son gonadales en su mayoría (15). Para el caso de los TCG extragonadales, no se conoce con certeza el porqué de la ubicación no gonadal.
A lo anterior debe sumarse que los TCG representan una paradoja, porque están compuestos de células pluripotenciales (en la década de 1970 se demostró pluripotencia en células de carcinoma embrionario) (16) y las células que lo forman tienen una ruta de proliferación limitada.
Para explicar dichos interrogantes, existen varias hipótesis, aun cuando se destacan dos: por un lado, la teoría de las células germinales explica que la ubicación extragonadal se genera por una migración anómala de las células germinales primordiales en la edad fetal, que se ubican en la línea media, donde sufren una transformación neoplásica; según la teoría, las células no son pluripotentes, pero tienen capacidad de reprogramación. Por otro, la teoría de las células madre pluripotentes plantea que el germinoma proviene de células germinales primordiales, pero los NGGCT provienen de células madre embrionarias que conservan su pluripotencia (17,18).
Otra forma de entender la pluripotencia de los TGC sugiere que las células germinales detienen su desarrollo embrionario temprano, evadiendo la diferenciación a gametocitos y manteniendo su pluripotencia, presumiblemente debido a señales somáticas incorrectas; luego permanecen inactivas hasta la pubertad, momento en el que por acción hormonal recuperan la actividad proliferativa y generan el tumor (19).
Germinoma
El germinoma es el más frecuente de los TCGI. Aparece, sobre todo, entre los 10 y los 12 años de edad. La mayoría se ubican a lo largo de la línea media y afectan la glándula pineal (65 %) y la región supraselar (30 %). En casos más raros, afecta ambas regiones (bifocales), el tálamo o los ganglios basales (20).
La diseminación del tumor no es muy frecuente; tienden a diseminarse mediante el líquido cefalorraquídeo, generalmente a la médula espinal, lo que, en general, le confiere buen pronóstico, con tasas de supervivencia de hasta un 90 % (21,22).
Genética
Las mutaciones en las vías KIT/RAS y AKT/mTOR y la amplificación del isocromosoma 12p se destacan dentro de los procesos genéticos que se asocian con la aparición del germinoma (23). Este tiene una particularidad respecto a otros TCG y a cualquier neoplasia maligna conocida: presenta baja metilación del ADN en todo el genoma. Se ha visto que la baja metilación conduce a la inestabilidad de los cromosomas (24). También se han hallado cambios en dos vías vinculadas con la proliferación, diferenciación, migración y apoptosis de células pluripotenciales embrionarias: MAPK y PI3K (25,26). Con todo, se ha concluido que la tríada genética propia del germinoma es baja metilación del ADN, inestabilidad severa de los cromosomas y alteraciones en las vías MAPK-PI3K (24). Véase la figura 1.
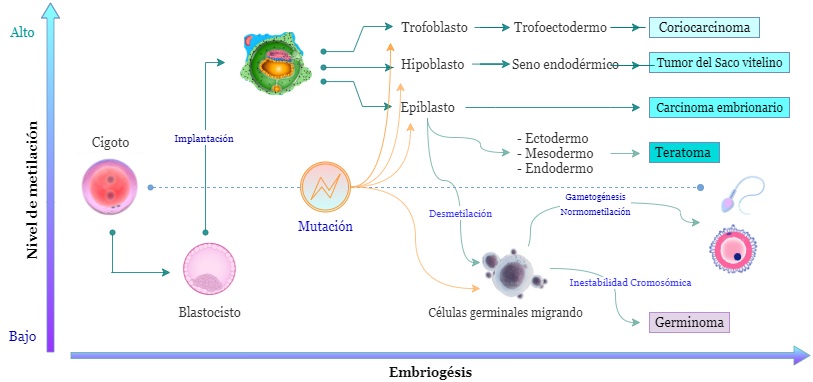
Figura 1. Genética y embriogénesis de los tumores de células germinales intracraneales. Del blastocisto con disco trilaminar se originan células primitivas, cuyas mutaciones específicas asociadas con hipermetilación del ADN favorecen la aparición de tumores de células germinales no germinomatosas. Las células germinales con baja metilación del ADN, sumado a inestabilidad cromosómica y mutaciones específicas, evolucionarán a germinomas. La normometilación del ADN, libre de mutaciones, dará paso a la gametogénesis.
Presentación histológica
Macroscópicamente, los germinomas son tumores blandos, con una superficie externa lisa, frágiles y color gris-rosado (27). Microscópicamente, están compuestos por dos componentes celulares que, formando lóbulos y láminas, se separan por bandas septales de tejido conectivo muy vascularizado. El primer componente es una población de células neoplásicas redondas, grandes, con un citoplasma claro, núcleos y nucléolos redondos y prominentes. Las mitosis son frecuentes (27). El segundo es un infiltrado inflamatorio reactivo no neoplásico compuesto principalmente por linfocitos T (28).
En ocasiones, el contenido inflamatorio es mayor, y ello lleva a posibles errores diagnósticos. Para estos casos, actualmente se usa la inmunohistoquímica con anticuerpos contra el antígeno OCT3/4, que hace parte de una familia de antígenos propios de las células germinales primordiales (29). Puede haber zonas de calcificación, generalmente en la periferia del tumor, y, a veces, granulomas (30).
Presentación clínica
El cuadro clínico generado por los germinomas dependerá de su tamaño y ubicación. Usualmente, los de la región pineal presentan síntomas por el aumento de la presión intracraneal, resultado de la hidrocefalia obstructiva (31). Son comunes las anomalías oftalmológicas, como pérdida de la agudeza visual y visión borrosa, que pueden deberse a compresión o invasión de la vía óptica (32). Son la principal causa del síndrome de Parinaud en jóvenes, que afecta los movimientos oculares extrínsecos (parálisis de la mirada hacia arriba, nistagmo convergente o retracción del párpado) e intrínsecos (midriasis), y produce, en pocas ocasiones, tinnitus y pérdida auditiva (33). En raras ocasiones hay cambios endocrinos y del desarrollo sexual, convulsiones y ataxia (34).
En la región supraselar, a menudo, tienen una presentación sintomatológica más lenta (34). Los síntomas principales son cefalea, náuseas, vómitos, letargo y trastornos visuales. Los últimos se presentan, generalmente, como hemianopsia temporal bilateral, diplopía, papiledema y atrofia del nervio óptico. Otras presentaciones comunes son la diabetes insípida y la disfunción del eje hipotálamo-hipófisis, manifestada como alteraciones del crecimiento y pubertad precoz (35).
Cuando el tumor aparece en los ganglios basales, genera síndrome piramidal por lesión de la cápsula interna. Las lesiones talámicas que afectan las conexiones con la corteza frontal pueden causar trastornos mentales caracterizados por desinhibición, inquietud, pobre introspección y euforia (36). En casos menos comunes, pueden presentarse episodios depresivos atípicos (37).
Diagnóstico
Un diagnóstico correcto es el precedente más importante para establecer un régimen de tratamiento adecuado. Para todos los TCG en el SNC, el diagnóstico se basa en una evaluación detallada de los signos y síntomas clínicos, en el análisis de biomarcadores y en el estudio de las neuroimágenes y de la histopatología.
Los biomarcadores más importantes son la subunidad β de la gonadotropina coriónica humana (β-hCG), la α-feto-proteína (AFP) y, en menor medida, la fosfatasa alcalina placentaria. Estos biomarcadores se pueden medir tanto en el líquido cefalorraquídeo como en la sangre, aun cuando la del líquido cefalorraquídeo es la medición con mayor sensibilidad y especificidad. El germinoma puro no secreta biomarcadores y, generalmente, es negativo para estos. Mediciones con concentraciones altas o moderadas indican que el germinoma está mezclado con otro tipo de TCGI (38). Cantidades bajas de β-hCG en germinomas puros indican diseminación leptomeníngea o presencia de células gigantes sincitiotrofoblásticas (39). En ese sentido, en Europa y Estados Unidos consideran secretor a un TGCI con valores de β-hCG ≥ 50 UI/l y AFP ≥ 10 ng/dl, o por encima de los valores que el laboratorio considere normales, en líquido cefalorraquídeo o suero (40).
Mediante los estudios de neuroimagen no se puede diferenciar el tipo de tumor intracraneal, por lo que el diagnóstico requiere confirmación histopatológica. Sin embargo, en la mayoría de los casos, por la profundidad de las lesiones y la complejidad de las áreas encefálicas afectadas, la realización de la biopsia puede aumentar la morbimortalidad. Por lo tanto, se han buscado hallazgos patognomónicos de germinoma en la tomografía axial computarizada de cráneo y en la resonancia magnética (RM) cráneo-espinal. Se prefiere la RM, porque, a diferencia de la tomografía, permite la caracterización intratumoral y la visualización de hemorragias (41).
En la RM, la mayoría de germinomas de la región pineal se muestran de hipointensos a isointensos en T1, e isointensos a hiperintensos en T2, marcadamente homogéneos; además, suelen tener un margen bien definido. A menudo, el germinoma de la región supraselar tiene margen mal definido; casi siempre muestra necrosis, quistes y hemorragia. También se muestran hipointensos a isointensos en T1 e isointensos a hiperintensos en T2, marcadamente heterogéneos. Por lo general, los germinomas de los ganglios basales y tálamo se muestran isointensos, en su mayoría presentan quistes intratumorales que se ven hipertensos respecto al líquido cefalorraquídeo, necrosis y hemorragia (41,42,43). Ninguno de estos hallazgos es patognomónico de germinoma, pero son predictores fiables de la extensión y multiplicidad de la lesión (44).
La verificación histopatológica debe realizarse mediante biopsia; sin embargo, por las adversidades que tiene su ejecución, está indicada en casos puntuales y se omite su realización en otras situaciones (40):
Si el resultado de biomarcadores es compatible con un tumor secretor, no está indicada la realización de biopsia, ya que se considera un tumor de células germinales no germinomatosas y debe iniciarse esquema terapéutico cuanto antes.
Si el resultado de biomarcadores es compatible con un tumor no secretor, está indicado realizar la biopsia, ya que varios tipos de TCGI pueden generar este patrón y tener comportamientos clínicos y pronóstico diferentes, entre ellos el germinoma y el teratoma.
Si el resultado de biomarcadores es compatible con un tumor no secretor, pero en la RM se evidencia extensión bifocal, en región pineal o supraselar, no está indicada la biopsia, pues se considera un germinoma.
Debido a que los germinomas tienen muy buena respuesta al tratamiento radio y quimioterapéutico, desde hace más de dos décadas la cirugía se ha relegado casi únicamente a ser un método diagnóstico (45). Existen diversos métodos quirúrgicos para tomar muestras óptimas para el estudio histopatológico: estereotácticamente, transesfenoidalmente y mediante craneotomía frontotemporal, aun cuando se prefiere la biopsia estereotáxica para su estudio intraoperatorio (46). Con poca confiabilidad, se puede evitar la biopsia mediante un estudio citológico de líquido cefalorraquídeo obtenido por punción lumbar (47).
La tinción inmunohistoquímica c-kit (CD 117) ha sido considerada un marcador útil en el diagnóstico de germinoma. Más recientemente, se ha informado que anticuerpos más nuevos, como OCT3/4 y SALL4 son más confiables (47).
Tratamiento
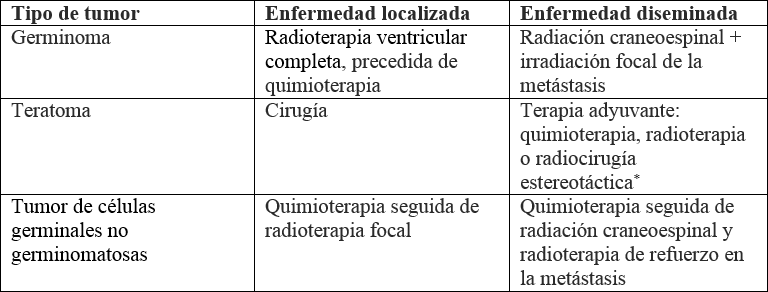
El germinoma muestra muy buena respuesta a la radioterapia. Se han informado tasas de supervivencia a cinco años de más del 90 % usando solo la radiación. Sin embargo, dosis altas y prolongadas mostraron alta toxicidad, por lo que se han estudiado estrategias de reducción del campo de radiación o de la dosis (48).
En la actualidad, el campo preferido para pacientes con germinoma diseminado es la irradiación craneoespinal (CSI). Para el germinoma no diseminado, la técnica recomendada es una radioterapia de campo reducido, como la radioterapia de cerebro completo o la radioterapia ventricular completa, para minimizar la toxicidad (48).
Tradicionalmente, los germinomas recibían al menos 50 Gy en el sitio primario del tumor sumado a CSI. Pero, al tener en cuenta los efectos tardíos: deterioro neurocognitivo, disminución del coeficiente intelectual, disfunción endocrina y efectos negativos en el desarrollo osteomuscular (48), se ha intentado reducir la dosis sin dejar de lado la contundencia de la terapia, cuya pérdida puede acarrear la recurrencia del cáncer.
Un estudio recientemente publicado demostró que dosis menores de 40 Gy en el sitio primario del tumor o menores de 25 Gy en CSI mantienen la tasa de remisión sin aumentar la recurrencia. Recomienda, además, que la radioterapia sea precedida por cuatro ciclos de quimioterapia basada en cisplatino + bleomicina o carboplatino + etopósido, ya que se aumenta la tasa de supervivencia hasta en un 100 % (49). Otros estudios recomiendan utilizar cuatro ciclos de cisplatino o carboplatino + etopósido con posterior dosis de 24 Gy de radioterapia ventricular completa o radioterapia de cerebro completo, alternando con CSI (50,51).
Los germinomas bifocales son confusos a la hora de tratarlos, ya que se desconoce si su comportamiento y respuesta terapéutica corresponde a un tumor diseminado o a uno confinado. Se ha demostrado que el uso de CSI genera mayores tasas de supervivencia libres de progresión del tumor bifocal, respecto a los demás esquemas usados, por lo que se consideró al germinoma bifocal como una enfermedad diseminada y su tratamiento se sugiere manejar de tal manera (52).
Tumores de células germinales no germinomatosas
Los NGGCT constituyen un grupo de neoplasias localizadas principalmente en región pineal y supraselar, aunque en ocasiones menos comunes ocupan la fosa posterior y los ventrículos. Aunque la Organización Mundial de la Salud los clasifica en la categoría NGGCT, los teratomas, a menudo, se consideran una entidad separada.
En general, los NGGCT son tumores agresivos. Según la clasificación japonesa de tumores pediátricos, el coriocarcinoma, el tumor del saco vitelino y el carcinoma embrionario corresponden al grupo de pobre pronóstico. El teratoma inmaduro y el germinoma con células sincitiotrofoblásticas tienen un pronóstico intermedio. Y el germinoma puro con el teratoma maduro conforman el grupo de buen pronóstico (53).
Genética
Siguiendo la explicación genética de la aparición del germinoma, es posible llegar a la explicación de la aparición de los NGGCT. Con la mutación MAPK/PI3K, puede surgir una célula pluripotente mutada que, según la metilación de su ADN, podrá diferenciarse a germinoma o a un NGGCT. Mientras mayor sea la metilación, más cerca estará de ser NGGCT. Así, la alta metilación del ADN es su característica genética diferencial con el germinoma (54). En general, para todos los NGGCT, los desequilibrios cromosómicos frecuentes se han descrito en los cromosomas 1, 8, 12, 13, 18 y X (55).
Presentación histológica
Los teratomas contienen componentes de tejido derivados de las tres capas germinales (endodermo, mesodermo y ectodermo). Pueden ser maduros, con apariencia similar a los tejidos diferenciados de un adulto (hueso, cartílago, cabello, grasa, epitelio, músculo o tejido nervioso), su alta diferenciación les confiere buen pronóstico, o pueden ser inmaduros, y parecerse a tejidos fetales primitivos, frecuentemente tejido conjuntivo y neuroectodérmico (similar al tubo neural), También pueden ser malignos y benignos. Estos últimos, generalmente, son lobulados y bien definidos, y tener áreas quísticas; a diferencia de los malignos, que suelen ser completamente sólidos, con bordes mal definidos y mucho edema peritumoral (56,57).
El carcinoma embrionario, por lo general, crece en láminas sólidas y puede mostrar estructuras glandulares o papilares. Está compuesto por células poligonales grandes que se asemejan a las del disco germinal embrionario, con núcleos pleomórficos grandes superpuestos y nucléolos prominentes. La actividad mitótica es alta y las áreas necróticas se observan con frecuencia. Las células tumorales expresan citoqueratinas, OCT3/4, fosfatasa alcalina placentaria y CD30 (58).
Los tumores del saco vitelino pueden mostrar una arquitectura diversa. El patrón reticular/microquístico se caracteriza por espacios quísticos revestidos por células neoplásicas dentro de un estroma mixoide. Otras presentaciones incluyen láminas sólidas, túbulos, glándulas, papilas o áreas con patrón hepatoide. El cuerpo de Schiller-Duval es un sello distintivo de este tipo de tumor y consiste en un vaso rodeado de células tumorales, en un espacio cístico. La inmunoexpresión del AFP es útil para el diagnóstico (59).
El coriocarcinoma muestra diferenciación trofoblástica. Puede estar formado por células citotrofoblásticas mononucleadas, con un citoplasma claro, cuyos núcleos son redondos y con un nucléolo prominente. O por sincitiotrofoblásticas grandes multinucleadas, con núcleos oscuros (60).
Presentación clínica
Los signos y síntomas dependen de las zonas encefálicas afectadas y son, en su mayoría, neurológicos. Entonces, su presentación es similar al germinoma. Algunos hallazgos son propios de algunos subtipos específicos de NGGCT, ya que no están relacionados con afectación intracraneal, sino con el comportamiento propio de las células tumorales, como es el caso de la pubertad precoz, que se presenta como efecto secundario de las altas concentraciones de h-CG (61).
En su presentación más habitual, los NGGCT pueden generar vómitos, cefalea, diplopía y otros síntomas consecuentes a la hidrocefalia obstructiva y a la implicación de las vías oculares. Tras la afectación hipofisaria, sobre todo por teratomas, el hipocortisolismo y el hipotiroidismo son los déficits más frecuentes (62).
Diagnóstico
El abordaje diagnóstico de los NGGCT sigue las mismas pautas y estudios paraclínicos e imagenológicos que el germinoma. Empero, los resultados de estos otorgan hallazgos distintivos que permiten hacer el diagnóstico diferencial. El más importante de ellos lo brindan los biomarcadores tumorales, que suelen ser positivos para los NGGCT. En la tabla 1 se muestra el comportamiento esperado de los biomarcadores para cada tipo de TCGI (53).
Tabla 1. Patrón de biomarcadores tumorales por subtipo de tumor de células germinales intracraneales
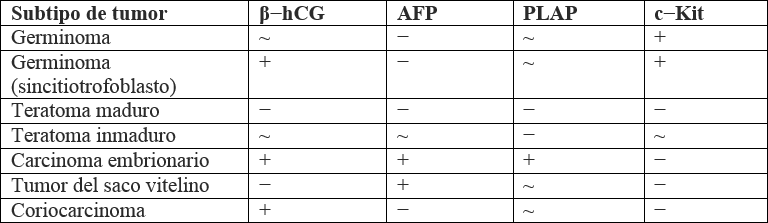
β-hCGsubunidad β de la gonodatropina coriónica humana
AFPα-feto-proteína
PLAPfosfatasa alcalina placentaria
+positivo
-negativo
~indeterminado
Las imágenes diagnósticas, como se ha dicho, muestran características similares para todos los TCGI. Algunas particularidades permiten descartar los germinomas como diagnóstico y pensar en NGGCT: múltiples zonas hemorrágicas y gran heterogeneidad tumoral (63). Los teratomas maduros suelen mostrar quistes grandes; mientras que los inmaduros muestran quistes pequeños. El tumor del saco vitelino, a menudo, tiene forma irregular (64).
En general, los NGGCT pueden diagnosticarse en función de la elevación en suero o líquido cefalorraquídeo de los marcadores tumorales AFP y β-hCG, respectivamente, con o sin confirmación histológica (65).
Tratamiento
Los teratomas, maduros e inmaduros, cimientan su tratamiento en la cirugía resectiva tumoral, ya que son insensibles a la quimio y radioterapia. En aquellos pacientes que no se logra la resección total, se debe iniciar manejo coadyuvante con radioterapia focal, quimioterapia o radiocirugía estereotáctica (66).
El resto de los NGGCT son radiosensibles, pero la supervivencia a los 5 años tras la CSI estándar es pobre (25 %-45 %) (53,67). Por ello, aunque el régimen de tratamiento sigue sin estar claro y se mantiene en estudio, actualmente la Sociedad Europea de Oncología Pediátrica recomienda quimioterapia seguida de resección quirúrgica, si hay enfermedad residual o radioterapia focal (54 Gy en 30 fracciones). Los agentes antineoplásicos usados incluyen carboplatino, etopósido, bleomicina, ifosfamida y vinblastina en diversas combinaciones (tabla 2). Al paciente con enfermedad metastásica debe ofrecérsele quimioterapia seguida de CSI (30 Gy en 20 fracciones) y radioterapia de refuerzo (24 Gy en 15 fracciones) en sitios primarios y metastásicos (67).
Conclusiones
Los TCGI se subclasifican en germinoma y NGGCT. Genéticamente, los germinomas deben su existencia a hipometilación del ADN, y los NGGCT, a hipermetilación. El germinoma, histológicamente, está formado por un estrato neoplásico de células grandes y redondas con núcleos prominentes. Los NGGCT presentan altas tasas mitóticas y atipia, lo cual les confiere peor pronóstico.
El diagnóstico se basa en la medición de biomarcadores, que suelen ser negativos para el germinoma, y positivos para los NGGCT. Las neuroimágenes no ofrecen mayor información diferencial, y la confirmación es histológica mediante biopsia o estudio citológico del líquido cefalorraquídeo.
El tratamiento varía según el tipo de tumor y la gravedad de la enfermedad: en germinomas localizados, se basa en radioterapia ventricular completa de baja intensidad; en teratomas, por ser quimio y radiorresistentes, se basa en la resección quirúrgica. Entre tanto, los NGGCT, al tener peor pronóstico, requieren quimioterapia como primera línea.

