











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El cáncer es un término genérico empleado para definir a un grupo de enfermedades caracterizadas por el crecimiento y diseminación descontrolada de células anormales, que pueden afectar cualquier parte del organismo y que sin un tratamiento adecuado pueden provocar la muerte. Aunque las causas del desarrollo del cáncer no han sido completamente dilucidadas, se sabe que numerosos factores aumentan el riesgo, pues actúan de manera simultánea para iniciar y/o promover su crecimiento, incluidos muchos que son potencialmente evitables, como el consumo de tabaco, alcohol, el exceso de peso corporal y el sedentarismo, y otros que no lo son, por ejemplo, las mutaciones genéticas y las afecciones virales, entre otros factores. El cáncer es una de las principales causas de muerte en todo el mundo y representó casi diez millones de muertes en 2020 [1]. La proyección de la Sociedad Americana del Cáncer estima que para 2023 se reportarán 1.958.310 nuevos casos, con un número de fallecidos en el orden de los 609.820. El tipo de cáncer que sufrirá un mayor incremento será el cáncer de próstata, con un 3 %, después de haber tenido una disminución en estas últimas dos décadas [2].
Se estima que existen dos causas del desarrollo anormal en cánceres humanos: la primera es la alteración de las vías de señalización en las células y la segunda es la interrupción del ciclo celular [3], [4]. La farmacorresistencia es considerada otra de las principales dificultades en la terapia del cáncer; puede desarrollarse en todos los tipos de cáncer y en estrategias terapéuticas como la inmunoterapia, la terapia molecularmente dirigida y la quimioterapia [5]. Son muchos los esfuerzos que se han hecho para reducir los efectos adversos durante la terapia contra el cáncer, entre ellos, la prevención de los efectos secundarios en las células y tejidos cercanos, el aumento de la acumulación de fármacos y la eficacia en la lesión, así como el desarrollo de nuevos sistemas de administración y focalización de fármacos [6]. Para alcanzar estos objetivos, un proceso que ha recibido gran atención en estos últimos tiempos es el relacionado con el "reposicionamiento", que consiste en encontrar nuevas indicaciones terapéuticas para los medicamentos utilizados clínicamente en el tratamiento de patologías diferentes al cáncer [7]-[11]. Otra estrategia alternativa para el desarrollo de nuevos fármacos es el enfoque de hibridación, que ha demostrado ser exitoso para el desarrollo de agentes biológicamente eficaces [12], [13]. El término "fármaco híbrido" generalmente se refiere a la combinación de dos o más moléculas biológicamente activas o dos grupos farmacofóricos de diferentes compuestos en una molécula. La combinación da como resultado una nueva molécula, ahora conocida como híbrida, que puede ser más potente, menos potente o idéntica a su compuesto precursor. En las moléculas hibridas, un conector puede estar presente (híbridos con enlazadores) o no (híbridos fusionados), o pueden estar fusionados (quiméricos) [14]-[19].
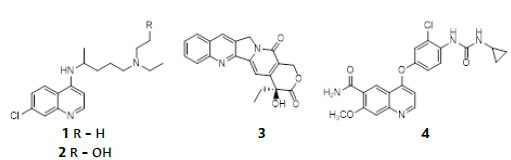
Como fragmento privilegiado, la quinolina es una molécula plana rígida, que se comporta como un farmacóforo presente en el núcleo de numerosos agentes fisiológicamente activos que muestran interesantes propiedades terapéuticas; por ejemplo, la cloroquina (CQ) 1, hidroxicloroquina (HCQ) 2, camptotecina 3, levatinib 4 [20]-[23] (Figura 1). Del mismo modo, cuando las quinolinas están constituidas en su estructura, con heteroátomos como el azufre, han resultado ser efectivas en modelos de enfermedades en donde su mecanismo de acción se ha asociado con la inhibición de la formación de ß-hematina, inhibición de la adhesión, migración, e invasión celular, inducción de apoptosis, estrés oxidativo, por su acción antipalúdica y antituberculosa, como agentes hipocolesterolémicos, y por su actividad antiproliferativa contra células cancerosas [24]-[32].
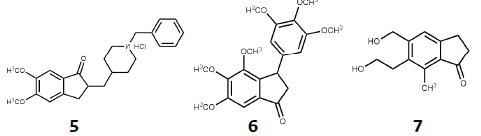
Otro núcleo privilegiado está constituido por la indanona, que en combinación con aldehídos genera las arilidenindanonas, que son consideradas como las primas rígidas de las chalconas y se generan incorporando al sistema αβ-insaturado cetónico de chalconas un anillo cíclico de cinco miembros. Generalmente son sintetizadas a partir de 1-indanona y benzaldehídos por medio de una reacción aldólica, por ejemplo, los compuestos 5 y 6 con actividad para el tratamiento de la enfermedad de Alzheimer y antineoplásicos, respectivamente [33], [34]. En la naturaleza encontramos algunas indanonas como la pterosina P 7 aislada del helecho de la especie Pteridium aquilinum [35]-[37], con actividad como antibacteriano, antiespasmódico, citotóxica, entre otras actividades [38], [39] (Figura 2).
En este trabajo presentamos el diseño y síntesis de nuevas moléculas que busca optimizar aún más los agentes anticancerígenos basados en la CQ; en ellas se modificó selectivamente la cadena lateral de la funcionalidad cuatro amino con un grupo que contiene azufre y la incorporación de indanonas metoxisustituidas (Figura 1). Seleccionamos este enfoque fundamentándonos en el hecho de que a los análogos de CQ sustituidos por 4-sulfanilindanonas aún no les ha sido explorada en profundidad su actividad anticancerígena. Se pudo observar que algunos de ellos muestran actividad antiproliferativa más efectiva que la CQ y la indanona individualmente; en el estudio de su potencial mecanismo de acción se pudo observar que existe una acumulación significativa de células en fases G2/M, que se acompañó de un aumento de células con G0/G1, y una disminución de S, ADN y contenido de ARN. Además, los compuestos indujeron distintos valores sub-G1, que representan la población de células apoptóticas y muertas.
Materiales y métodos
Los puntos de fusión se determinaron en un fusiómetro Fisher-JohnsMR y no están corregidos. La cromatografía en capa fina (CCF) se llevó a cabo en placas MerckMR silica F254 de 0,255 mm y las manchas se visualizaron mediante fluorescencia UV a 254 nm. Los espectros IR se determinaron mediante un espectrofotómetro Perkin-Elmer-MR Spectrum two y se expresan en cm-1. Los espectros de RMN de 1H y 13C se realizaron usando un espectrómetro JEOL EclipseMR 270 (a 270 MHz para espectros de 1H y 67,9 MHz para espectros de 13C), usando CDCl3 como solvente, y se reportan en ppm empleando como referencia el CHCl3 residual (δ 7 ,25 para RMN 1H y 77,0 para RMN 13C, respectivamente). Los reactivos químicos se obtuvieron de Aldrich Chemical CoMR, EE.UU. de Norteamérica. Todos los disolventes se destilaron y secaron de la manera habitual. El intermediario 8 ha sido reportado previamente [40].
Procedimiento general para la obtención de los derivados 2-{3-[(7-cloroquinolin-4-il) sulfanil] -1-hidroxi-propil}-indanosustitu ida-2,3-d ih idro -1H-inden-1-ona (11a-e)
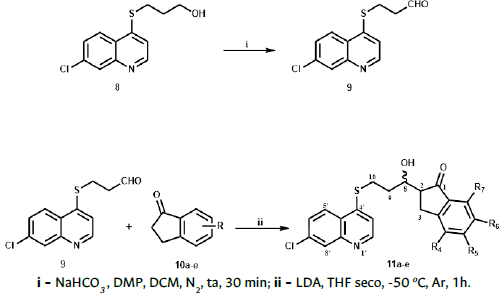
Para la obtención del aldehído 9, se disolvió el intermediario 8 (1 Eq) en 20 mL de diclorometano (DCM) seco, se agregó 10 Eq de bicarbonato de sodio (NaHCO3), 1,2 Eq de reactivo de Dess-Martin (DMP), en 5 mL DCM seco; la mezcla se mantuvo con agitación constante por 30 min a temperatura ambiente y atmósfera de N2. Finalizada la reacción, se lavó con solución saturada de Na2S2O3 (4x10 mL), solución saturada de NaHCO3 (2x10 mL), y con agua destilada (2x10 mL). La fase orgánica se secó con Na2SO4 anhidro, se filtró y el solvente se eliminó a presión reducida [41]. Debido a su inestabilidad, el compuesto 9 se usó sin previa caracterización.
Paralelamente, se procedió a sintetizar la diisopropilamina de litio (LDA), que se obtuvo al hacer reaccionar diisopropilamina seca 1 Eq (la diisopropilamina se secó con hidruro de calcio a 120 °C y posterior destilación a presión normal) y n-butillitio 1 Eq (disuelto en tolueno 1,6 M), a -78 °C, en atmósfera de Ar y agitación continua por 20 min. Se agregó gota a gota, por medio de una inyectadora, la indanona respectiva 10a-e (0,85 Eq) disuelta en 2 mL de THF seco en un tiempo de 15 min; seguidamente se adicionó el aldehído 9 disuelto en 1 mL de THF seco; la mezcla de reacción se agitó por 30 min hasta que la temperatura alcanzó los -50 °C [42]. La reacción se monitoreó por CCF; finalizada la reacción se adicionó al balón una solución saturada de cloruro de amonio (NH4Cl, 10 mL), además de una mezcla de acetato de etilo AcOEt:agua (1:1) (15 mL). La fase orgánica se separó y se trató con Na2SO4 anhidro; se filtró y evaporó el solvente a presión reducida. El sólido obtenido se purificó utilizando cromatografía de columna, con mezclas de ciclohexano:acetato de etilo (Cy:AcOEt); se obtuvo una mezcla de diasteroisómeros con rendimientos que van desde 55 % a un 75 % [43], [44].
(+/-)2-{10-[(7-cloroquinolin-4-il)sul/anil]-8-hidroxipropil}-2,3-dihi-dro-1H-inden-1-ona 11a
Sólido cristalino blanco. Purificación por columna: 100% Cy, 85:15, 75:25, 65:35, 3:2 Cy:AcOEt. Rendimiento: 75%. Punto de fusión: 173 - 175 °C. Infrarrojo (cm1): 3406 (-OH), 2923 (C-H alifática), 1691 (C=O). RMN 1H (270 MHz, CDCl3) δ ppm: 2,02-2,09 (m, 4H, H9); 2,71-2,88 (m, 2H, H2); 3,17-3,42 (m, 8H, H3, 10); 3,98-4,04 (m, 1H, H8); 4,54-4,57 (m, 1H, H8); 4,73 (sa, 1H, OH);7,17 (d, 1H, H3', J = 4,94 Hz); 7,26 (d, 1H, H3', / = 4,94 Hz); 7,31-7,48 (m, 6H, H4, 6, 6'); 7,60 (d, 1H, H6, J = 7,18 Hz); 7,71 (d, 1H, H7, J = 7,67 Hz); 7,76 (d, 1H, H7, / = 7,67 Hz); 7,98 (d, 1H, H5', / = 8,91 Hz); 7,98-8,02 (m, 3H, H5', 8' x 2); 8,65;8,69 (d, 1H, H2', J = 4,94 Hz). RMN 13C (68 MHz, CDCl3) δ ppm: 27,0(C3 ó 10); 27,6(C3 ó 10); 27,9(C3 ó 10); 29,8(C3 ó 10); 33,4(C9); 34,5(C9); 51,7(C2); 53,2(C2); 69,6(C8); 71,4(C8); 116,2(C3'); 123,9(C7); 124,2(C7); 125,0(C5' ó 8'); 125,1(C5' ó 8'); 126,6(C6' ó 4 ó 6); 126,7(C6' ó 4 ó 6); 127,3(C6' ó 4 ó 6); 127,3(C6' ó 4 ó 6); 127,6(C6' ó 4 ó 6); 127,9(C6' ó 4 ó 6); 128,9(C5' ó 8'); 135,2(C5); 135,6(C5); 135,8: 135,8; 136,4; 137,1; 147,9; 147,9; 148,0; 150,2(C2); 150,3(C2); 153,5; 153,4; 207,6(C1); 209,6(C1). F.M: C21H18ClNO2S, P.M: 383,89 g/mol.
(+/-)2-{10-[(7-cloroquinolin-4-il)sul/am'l]-8-hidroxipropil}-5-me-toxi-2,3-dihidro-1H-inden-1-ona 11b
Sólido cristalino blanco. Purificación por columna: 100% Cy, 4:1,3:2, 1:1, 2:3, 3:7 Cy:AcOEt. Rendimiento: 65%. Punto de fusión: 158 - 160 C. Infrarrojo (cm-1): 3375 (-OH), 2925 (C-H alifática), 1677 (C=O), 1026 (ar C-O-C al). RMN 1H (270 MHz, CDCl3) δ ppm: 1,94-2,06 (m, 4H, H9); 2,65-2,86 (m, 2H, H2); 3,10-3,27 (m, 4H, H3); 3,35-3,41 (m, 4H, H10); 3,86 (s, 3H, OMe), 3,92-4,01 (m, 1H, H8); 4,46-4,53 (m, 1H, H8); 4,90 (sa, 1H, OH); 6,83-6,91 (m, 4H,
H4, 6); 7,18 (d, 1H, H3', J = 4,67 Hz); 7,22 (d, 1H, H3, / = 4,67 Hz); 7,45 (dd, 2H, H6', J = 1,97, 8,91 Hz); 7,62 (d, 1H, H7, J = 8,67 Hz); 7,68 (d, 1H, H7, J = 8,67 Hz); 7,97 (d, 1H, H5', J = 8,91 Hz); 7,998,02 (m, 3H, H5', 8' x 2); 8,65 (d, 1H, H2', J = 4,94 Hz); 8,68 (d, 1H, H2', J = 4,94 Hz). RMN 13C (68 MHz, CDCl3) δ ppm: 27,0(C3 ó 10); 27,7(C3 ó 10); 27,9(C3 ó 10); 29,8(C3 ó 10); 33,3(C9); 34,6(C9); 51,6(C2); 53,2(C2); 55,8(OMe); 69,8(C8); 71,6(C8); 109,7(C4 ó 6); 109,9(C4 ó 6); 115,7(C3'); 116,0(C4 ó 6); 116,2(C4 ó 6); 125,0(C5' ó 8'); 125,1(C5' ó 8'); 125,6(C7); 125,9(C7); 127,3(C6'); 127,3(C6'); 128,9(C5' ó 8'); 130,4; 135,7; 148,0; 150,2(C2); 150,3(C2); 156,6; 157,4; 165,9; 166,2; 205,7(C1); 207,3(C1); F.M: C22H20ClNO3S, P.M: 413,92 g/mol.
(+/-)2-{10-[(7-cloroquinolin-4-il)sulfanil]-8-hidroxipropil}-4,5-dime-toxi-2,3-dihidro-1H-inden-1-ona 11c
Sólido amarillo. Purificación por columna: 100% Cy, 95:5,9:1, 85:15, 4:1, 75:25, 7:3, 65:35, 3:2, 55:45, 1:1, 45:55, 2:3, 35:65, 3:7 Cy:AcOEt. Rendimiento: 60%. Punto de fusión: 130 - 132 C. Infrarrojo (cm-1): 3392 (-OH), 2927 (C-H alifática), 1693 (C=O), 1081 (ar C-O-C al). RMN 1H (270 MHz, CD3OD) δ ppm: 1,97-2,04 (m, 2H, H9); 2,20-2,27 (m, 2H, H9); 2,97-3,08 (m, 2H, H2); 3,23-3,25 (m, 4H, H3); 3,35-3,53 (m, 4H, H10); 3,88 (s, 3H, OMe); 3,94 (s, 3H, OMe); 4,18-4,21 (m, 2H, H8); 4,39-4,42 (m, 1H, OH); 7,12 (d, 2H, H6, J = 8,42 Hz); 7,45 (d, 2H, H7, J = 8,40 Hz); 7,75-7,77 (m, 4H, H3', 6'); 8,05 (d, 2H, H8',/ = 1,97 Hz); 8,31 (d, 2H, H5', J = 8,88 Hz); 8,77 (d, 2H, H2', J = 5,70 Hz). RMN 13C (68 MHz, CD3OD) δ p pm: 25,9(C3 ó 10); 27,9(C3 ó 10); 31,7(C9); 52,4(C2); 55,6(OMe); 59,3(OMe); 70,7(C8); 112,9(C6); 115,9(C3'); 119,9(C7); 122,7(C8'); 124,6; 125,6(C5'); 128,6(C6'); 130,9; 145,3; 147,1(C2'); 158,3;206,2(C1). FM: C23H22ClNO4S, P.M: 443,94 g/mol.
(+/-)2-{10-[(7-cloroquinolin-4-il)sulfanil]-8-hidroxipropil}-4,7-dime-toxi-2,3-dihidro-1H-inden-1-ona 11d
Sólido marrón. Purificación por columna: 100% Cy, 4:1, 7:3, 3:2, 55:45, 1:1, 4:3 Cy:AcOEt. Rendimiento: 69%. Punto de fusión: 143 - 145 °C. Infrarrojo (cm1): 3243 (-OH), 2921 (C-H alifática), 1695 (C=O), 1026 (ar C-O-C al). RMN 1H (270 MHz, CDCl3) δ ppm: 2,04-2,10 (m, 4H, H9); 2,62-2,83 (m, 2H, H2); 2,98- 3,15 (m, 4H, H3); 3,30- 3,46 (m, 4H, H10); 3,82 (s, 3H, OMe); 3,89 (s, 3H, OMe); 3,96-4,12 (m, 1H, H8); 4,40- 4,55 (m, 1H, H8); 4,94 (sa, 1H, OH); 6,68 (d, 1H, H5, J = 8,91Hz); 6,73 (d, 1H, H5, J = 8,91 Hz); 6,97 (d, 1H, H6, J = 8,64 Hz); 7,01 (d, 1H, H6, J = 8,64 Hz); 7,40-7,42 (m, 2H, H3'); 7,53 (dd, 2H, H6', J = 9,13 Hz); 8,06 (d, 2H, H5', J = 8,88 Hz); 8,27 (sa, 2H, H8'); 8,68 (d, 2H, H2', J = 5,43 Hz). RMN 13C (68 MHz, CDCl3) δ ppm: 24,3(C3 ó 10); 26,2(C3 ó 10); 27,3(C3 ó 10); 28,1(C3 ó 10); 33,2(C9); 34,3(C9); 51,5(C2); 53,2(C2); 56,0(OMe); 56,1(OMe); 69,8(C8); 71,4(C8); 109,7(C5); 110,1(C5); 115,5(C3'); 117,4(C6) 117,8(C6) 124,8(C5' ó 8'); 125,1(C5' ó 8'); 125,4(C5' ó 8'); 126,1(C5' ó 8') 128,6(C6'); 128,7(C6'); 143,9; 146,7(C2'); 150,3; 150,6; 151,8; 152,1; 207,3(C1). F.M: C23H22ClNO4S, P.M: 443,94 g/mol.
(+/-)2-{10-[(7-cloroquinolin-4-il)sulfanil]-8-hidroxipropil}-5,6-dime-toxi-2,3-dihidro-1H-inden-1-ona 11e
Sólido amarillo. Purificación por columna: 100% Cy, 4:1,7:3, 3:2, 55:45, 1:1, 45:55, 2:3, 35:65, 3:7 Cy:AcOEt. Rendimiento: 55%.
Punto de fusión: 142 - 143 C. Infrarrojo (cm-1): 3263 (-OH), 2923 (C-H alifática), 1687 (C=O), 1030 (ar C-O-C al). RMN 1H (270 MHz, CDCl3) δ ppm: 1,94-2,04 (m, 4H, H9) 2,61-2,73 (m, 1H, H2); 2,842,88 (m, 1H, H2); 3,04-3,28 (m, 4H, H3); 3,37-3,42 (m, 4H, H10); 3,84 (s, 3H, OMe); 3,89 (s, 3H, OMe;, 3,91-3,99 (m, 1H, H8); 4,464,52 (m, 1H, H8); 4,87 (sa, 1H, OH); 6,83 (s, 1H, H4); 6,87 (s, 1H, H4) 7,08 (s, 1H, H7); 7,14 (s, 1H, H7); 7,19 (d, 1H, H3', / = 4,70 Hz); 7,27 (d, 1H, H3', /= 4,70 Hz); 7,45 (dd, 2H, H6', J = 1,73, 8,91 Hz); 7,99 (d, 1H, H5', J = 8,88 Hz); 8,01-8,03 (m, 3H, H5', 8' x 2); 8,65 (d, 1H, H2', J = 4,70 Hz); 8,69 (d, 1H, H2', J = 4,70 Hz). RMN 13C (68 MHz, CDCl3) δ ppm: 27,0(C3); 27,5(C3); 27,9(C10); 29,6(C10);
33,3(C9); 34,7(C9); 51,8(C2); 53,2(C2); 56,2(OMe); 56,3(OMe); 69,9(C8); 71,6(C8); 104,4(C7); 104,6(C7); 107,4(C4); 107,6(C4); 116,2(C3'); 116,2(C3'); 125,0(C5'); 125,1; 127,3(C6'); 127,3(C6'); 128,9(C8'); 129,0; 129,8; 135,8; 135,8; 148,0; 149,1 149,8; 150,0; 150,1; 150,2(C2'); 150,2(C2'); 156,2; 156,5; 206,0(C1);207,6(C1). FM: C23H22ClNO4S, P.M: 443,94 g/mol.
Líneas celulares
Todas las líneas celulares fueron suministradas por la American Tissue Culture Collection (ATCC). La línea celular A549 es derivada de adenocarcinoma de pulmón. Las líneas celulares MRC-5 y BJ se utilizaron como control no tumoral y representan fibroblastos humanos. MRC-5 LD y BJLD son líneas celulares de fibroblastos de pulmón humano resistentes a la doxorrubicina. La línea CCRF-CEM se deriva de la leucemia linfoblástica T, con una alta quimiosensi-bilidad, mientras que K562 representa células de una muestra de paciente con leucemia mieloide aguda con translocación bcr-abl. La sublínea resistente a daunorrubicina de células CCRF-CEM (CEM-DNR bulk) y la sublínea resistente a paclitaxel K562-TAX fueron seleccionadas mediante el cultivo de líneas celulares maternas en concentraciones crecientes de daunorrubicina o paclitaxel, respectivamente. Las células CEM-DNR sobreexpresan MRP-1 y proteína P-glicoproteína, mientras que las células K562-TAX sobreexpresan solo la glicoproteína P. Ambas proteínas pertenecen a la familia de transportadores ABC y están implicadas en el fenómeno primario y/o adquirido de resistencia a múltiples fármacos. La línea celular U2OS se deriva de osteosarcoma, HCT116 es una línea celular tumoral colorrectal y su contraparte (HCT116p53-/-, Horizon Discovery Ltd, Reino Unido) es un modelo de cánceres humanos con mutación del gen p53 que frecuentemente es asociada con mal pronóstico [45], [46]. Las células se mantuvieron en matraces plásticos nunc/corning de 80 cm2 para cultivo de tejido y se cultivaron en un medio de cultivo celular de acuerdo con las recomendaciones ATCC u Horizon (DMEM/RPMI 1640 con 5 g/L de glucosa, 2 mM de glutamina, 100 U/mL; de penicilina, 100 mg/mL de estreptomicina, 10% de suero fetal de ternera y NaHCO3) [45]-[48].
Ensayo de citotoxicidad empleando MTS
La actividad de los compuestos se determinó utilizando 3-(4,5-di-metiltiazol-2-il)-5-(3-carboximetoxifenil)-2-(4-sulfofenil)-2H-te-trazolio (MTS) y se realizó en el Instituto de Medicina Molecular y Traslacional mediante una plataforma robótica (High-ResBioso-lutions). Las suspensiones celulares se prepararon y diluyeron de acuerdo con el tipo de célula en particular y la densidad celular objetivo esperado (25000-35000 células/mL según las características de crecimiento celular). Las células se añadieron mediante pipeta automática (30 μL) en placas de microtitulación de 384 pozos. Todos los compuestos ensayados se disolvieron en DMSO al 100% y se añadieron diluciones cuádruples de la concentración de ensayo prevista en alícuotas de 0,15 μL en el momento cero a los pozos de la placa de microtitulación mediante el manipulador de líquidos sin contacto eco-acústico Echo550 (LabcyteMR). Los experimentos se realizaron en duplicados y tres réplicas biológicas al menos. Las células se incubaron con los compuestos ensayados durante 72 h a 37 °C, en una atmósfera de CO2 al 5% al 100% de humedad. Al final del período de incubación, las células se analizaron mediante la prueba MTS. Las alícuotas (5 ßL) de la solución madre MTS se pipetearon en cada pozo y se incubaron durante 1-4 h adicionales. Después de este período de incubación, la densidad óptica (DO) se midió a 490 nm con un lector Envision (PerkinElmerMR). La supervivencia de las células tumorales (SCT) se calculó mediante la siguiente ecuación: SCT = (pozo expuesto a DOdrug/pozos DOcontrol medios) x 100%.
El valor de CI50, la concentración compuesta que es letal para el 50% de las células tumorales, se calculó a partir de las curvas de dosis-respuesta apropiadas en el software Dotmatics [45]-[48].
Análisis del ciclo celular y de la apoptosis
Las células CCRF-CEM se sembraron a una densidad de 1 x 106 células por mL en placas de seis pozos (TTP) y se cultivaron con compuestos en concentraciones correspondientes a 1 x ó 5 x valor CI50. Junto con las células tratadas con compuestos, se recolectó una muestra tratada con vehículo en el mismo punto de tiempo. Después de 24 h, las células se lavaron con solución salina tamponada con fosfato frío (PBS) y se fijaron en etanol al 70% añadido gota a gota y almacenado durante la noche a -20 C. Posteriormente, las células se lavaron en tampón de citrato hipotónico, se trataron con RNasa (50 ßg/mL) y se tiñeron con yoduro de propidio. Para la medición se utilizó citometría de flujo utilizando un láser de haz único de 488 nm (Becton DickinsonMR). El ciclo celular fue analizado por el software ModFitLT (VerityMR), y la apoptosis se midió en un modelo logarítmico que expresa el porcentaje de partículas con contenido de propidio inferior al de las células en la fase G0/G1 (<G1) del ciclo celular. La mitad de la muestra se utilizó para el marcado de anticuerpos pH3Ser10 (SigmaMR) y el posterior análisis de citometría de flujo de las células en mitosis [46].
Análisis de incorporación de BrDU
Las células se cultivaron y procesaron como se describe en el método anterior [45]-[48]. Antes de la cosecha, se agregó 5-bro-mo-2-deoxiuridina (BrDU) 10 μ M a las células para el marcado de pulsos durante 30 min. Luego, las células se lavaron con PBS y se fijaron con etanol frío al 70% de -20 °C y se almacenaron en un congelador durante la noche. Antes del análisis, las muestras se incubaron en hielo durante 30 min, se lavaron una vez con disolución salina tamponada con fosfato (PBS) y se volvieron a suspender en HCl 2 M durante 30 min a temperatura ambiente (Ta) para desnaturalizar su ADN. Después de la neutralización con una solución de Na2B4O7 (bórax) 0,1 M, las células se lavaron con PBS que contenía 0,5% de Tween-20 y 1% de albúmina de suero bovino (BSA). A continuación, se realizó la tinción con anticuerpo primario anti-BrDU (Exbio) durante 30 min a Ta en la oscuridad. Luego, las células se lavaron con PBS y se tiñeron con anticuerpos secundarios anti-ratón-FITC (SigmaMR) a Ta y en la oscuridad. Después de otro lavado con PBS e incubación con yoduro de propidio (0,1 mg/mL) y RNasa A (0,5 mg/mL) durante 1 h a Ta en la oscuridad, las células se analizaron mediante citometría de flujo utilizando un láser de haz único de 488 nm (FACSCalibur, Becton Dickinson™, Franklin Lakes, NJ, EE.UU.
Análisis de incorporación de BrU
Las células se cultivaron y trataron como se describió anteriormente [45]-[48]. Antes de la cosecha, siguió el etiquetado de pulsos con 1 mM de 5-bromouridina (BrU) durante 30 min. Las células se fijaron en paraformaldehído tamponado al 1% con NP-40 al 0,05% a Ta durante 15 min, y luego se almacenaron a 4 °C durante la noche. Antes de la medición, se lavaron con glicina al 1% en PBS, se lavaron con PBS nuevamente y se tiñeron con anticuerpos primarios anti-BrDU de reacción cruzada a BrU (Exbio) durante 30 min a Ta en la oscuridad. A partir de este punto, el experimento se realizó exactamente como en el método anterior.
Resultados y discusión
Síntesis
Para la síntesis de los derivados 11a-e, se realizaron varios intentos a fin de alcanzar el aldehído respectivo 9; entre los procedimientos empleados se mencionan diferentes agentes oxidantes, como el ácido 2-iodoxibenzoico (IBX), ácido 2-iodobenzoico estabilizado (SIBX), reacción de swern y el reactivo de Dess-Martin (DMP); sin embargo, el mejor resultado fue encontrado con el reactivo de DMP, con el cual se obtuvo el compuesto 9 (Figura 3); debido a su inestabilidad no fue posible su purificación, por lo que se usó sin caracterización previa en el siguiente paso de reacción. La reacción de condensación se realizó con diferentes indanonas 10a-e, empleando como base di-isopropilamina de litio (LDA), en tetrahi-drofurano (THF) seco, atmosfera de Ar, a una temperatura de -78 °C. La reacción se completa en un período de 50 min aproximadamente, con rendimientos que van desde 55 a 75%. Obtenida la mezcla de diasteroisómeros, se realizaron intentos para separar dicha mezcla, por medio de una columna cromatográfica, en donde se elevó progresivamente la polaridad de la fase móvil, pero no se obtuvo la separación deseada.
El compuesto 2-{10-[(7-cloroquinolin-4-il)sulfanil]8-hidroxipro-pil}5,6-dimetoxi-2,3-dihidro-1H-inden-1-ona (11e) se empleará como modelo para ilustrar la discusión. Este compuesto se obtuvo como una mezcla de diasteroisómeros con una fórmula molecular C23H22ClNO4S, como un sólido amarillo, soluble en DCM, su punto de fusión fue de 142-143 C y se obtuvo con un rendimiento de 55%. En el espectro de IR se observaron: una banda intensa a 3263 cm-1, correspondiente al estiramiento O-H, a 2923 cm-1 aparece una banda característica de estiramientos C-H de la zona alifática, a 1687 cm-1 una banda típica de carbono carbonílico C=O de cetona, y a 1030 cm-1 la banda correspondiente a los estiramientos C-O-C de los grupos metoxi presentes en la molécula. Cuando se procede a analizar el espectro de RMN 1H, se observa que varias señales aparecen duplicadas, lo que se atribuye a que el compuesto se encuentra bajo la forma de una mezcla de diasteroisómeros. El espectro nos muestra a campo alto un multiplete con un desplazamiento químico desde 1,94 a 2,04 ppm, que integra para 4H, que se asignó a los protones enlazados al carbono nueve; entre 2,61-2,73 y 2,84-2,88 ppm se observan dos multipletes que integran para 1H cada uno, asignado al protón sobre el carbono dos del anillo de la indano-na, entre 3,04-3,28 ppm se presenta un multiplete que integra para 4H, que fue asignado a los protones unidos al carbono tres de la indanona. Entre 3,37-3,42 ppm se tiene un multiplete que integra para 4H asignados a los protones sobre el carbono dies en la cadena alifática. A 3,84 y 3,89 ppm se observan dos singletes que integran para 6H cada uno, que fueron asignados a los grupos metoxi en las posiciones cinco y seis del anillo de la indanona. A 3,91-3,99 y 4,464,52 ppm se observan dos multipletes que integran para 1H cada uno, los cuales fueron asignados al protón del carbono ocho presente en la cadena alifática. Y, para finalizar, en la zona alifática se muestra un singlete ancho a 4,87 ppm, que se asigna al protón del grupo hidroxilo en el compuesto 11e. A campo bajo se presentan seis señales que se asignan a siete protones aromáticos. A 6,83 y 6,87 ppm se presentan dos singletes que integran cada uno para 1H, asignado al protón cuatro, y seguidamente, a 7,08 y 7,14 ppm, otros dos singletes que integran para 1H cada uno asignado al protón siete presente en el anillo de la indanona. En cuanto a los protones del anillo quinolínico, las señales se encuentran duplicadas, pero no se observa una diferencia significativa en lo que respecta a los desplazamientos y la multiplicidad, al ser comparada con los espectros de los compuestos quinolínicos previamente discutidos.
En el espectro de RMN 13C se confirma igualmente la presencia de los productos de condensación, con base en la presencia de cuatro señales a 27,0 y 27,5 ppm, que se asigna al carbono tres del anillo de la indanona, a 51,8 y 53,2 ppm el carbono dos, a 69,9 y 71,6 ppm las señales correspondientes al carbono ocho y en 206,0 y 207,6 ppm las señales correspondientes al C carbonílico de la cetona. Todas estas señales se encuentran duplicadas por presentar una mezcla de diasteroisómeros en la muestra. En el espectro se observan 38 señales totales pertenecientes a los carbonos no equivalentes en el compuesto 11e, dentro de lo que cabe mencionar que dos corresponden con los carbonos metílicos de los grupos metoxi, seis se corresponden con carbonos metilénicos, 16 con carbonos metínicos y 14 señales que indican el número de carbonos cuaternarios. A 27,9 y 296 ppm se muestran dos señales asignadas al carbono diez. A 33,3 y 34,7 ppm se observan las señales asignadas al carbono nueve. A 56,2 y 56,3 ppm se tienen las señales de los grupos metoxi que se encuentran unidos en las posiciones cinco y seis del anillo de la indanona. En lo que respecta a la zona aromática se evidencian dos señales en 104,4 y 104,6 ppm asignada al carbono siete y a 107,4 y 107,6 ppm se tienen dos señales que son asignadas al carbono cuatro, correspondientes al anillo de la indanona. Las señales asignadas a los protones metínicos del anillo quinolínico no presentan diferencias significativas en los desplazamientos cuando se comparan con los del compuesto reportados previamente. En cuanto a las diferencias de los desplazamientos de los carbonos en la estructura de los derivados 11a-e, se observa diferencia en los desplazamientos y multiplicidad que se atribuye al patrón de sustitución presente sobre el anillo aromático de la indanona. Estas asignaciones inequívocas se realizaron apoyándonos además en el análisis de los espectros DEPT 135°, COSY, HETCOR.
Actividad citotóxica
La actividad citotóxica in vitro de los compuestos finales 11a-e se evaluó después de tres días de incubación frente a ocho líneas celulares derivadas de tumores sólidos humanos, incluidos carcinomas de pulmón (células A549) y colon (HCT116 y HCT116p53-/-), así como líneas celulares de leucemia (CCRF-CEM, CEM-DNR, K562 y K562-TAX) y osteosarcoma (células U2OS). A modo de comparación, se usaron líneas celulares resistentes a la doxorrubicina de fibroblastos BJ y MRC-5 no malignos y de fibroblastos BJLD y MCR-5LD. Las concentraciones que inhiben el crecimiento celular en un 50% (CI50) se determinaron mediante una tinción metabólica cuantitativa con 3-(4,5-dimetiltiazol-2il)-5-(3-carboximetoxifeni-l)-2-(4-sulfofenil)-2H-tetrazolio (MTS) y se resumen en la Tabla 1.
Tabla 1 Actividad citotóxica (CI50 , μM) de derivados de 11a-e después de 72 h de incubación con líneas celulares humanas cancerosas y no cancerosas.
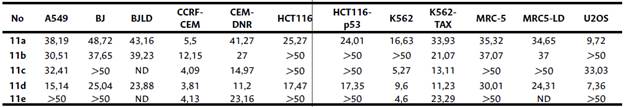
A549: adenocarcinoma de pulmón, MRC-5 y BJ: células no tumorales de fibroblastos humanos, MRC-5 LD y BJLD: fibroblastos de pulmón humano resistentes a la doxorrubicina, CCRF-CEM: leucemia linfoblástica T, K562: leucemia mieloide aguda con translocación bcr-abl, CCRF-CEM: sublínea celular de leucemia resistente a daunorrubicina, K562-TAX: sublínea celular de leucemia mieloide aguda resistente a paclitaxel, U2OS: línea celular de osteosarcoma, HCT116: línea celular tumoral colorrectal, HCT116p53-/-: línea celular tumoral colorrectal con mutación en el gen p53.
Los resultados mostraron que la línea celular CCRF-CEM fue la más sensible a los derivados de 11a, c-d. Con la excepción del compuesto 11d con un valor de CI50 en el rango de 11,2 /.μM, los compuestos fueron menos activos contra sus homólogas CEM-DNR resistentes a la daunorrubicina. Con algunas excepciones, en el caso de las células K562 y la correspondiente línea celular K562-TAX resistente a taxol, se observó una diferencia significativa para el derivado 11b con un valor dos veces mayor contra K562-TAX que la línea celular K562. Por otro lado, con la excepción de 11b y d los compuestos 11a, c y e fueron en el rango de dos y cinco veces más potentes contra K562 que la línea celular resistente K562-TAX. Estos resultados indican que otros mecanismos, además de la glicoproteína P, que es común para ambas líneas celulares, son responsables de la resistencia.
A excepción del derivado 11d, el resto de los compuestos pueden considerarse inactivos frente al adenocarcinoma de pulmón humano A549 (CI50 > 1 5 μM). La actividad citotóxica de todos los compuestos probados contra las líneas celulares de carcinoma de colon (HCT116 y HCT116p53-/-) fue similar, poco activos. Se observó un ligero aumento en la actividad citotóxica de los compuestos 11a y d frente a células cancerosas humanas de osteosarcoma (U2OS) (CI50 9,72 y 7,36 /¿M), respectivamente. Los compuestos 11a-e se pueden considerar como no tóxicos en las líneas celulares no cancerosas BJ, MRC-5, BJLD y MRC-5LD.
A partir de estos resultados, es obvio, con algunas excepciones, que el derivado 11d exhibió una citotoxicidad selectiva más pronunciada (en comparación con las células BJ y MRC-5) contra las células cancerosas humanas de HCT116 (cáncer colorrectal humano con p53 de tipo salvaje) y HCT116p53-/- (cáncer colorrectal humano con p53 eliminado), así como líneas celulares de leucemia (CCRF-CEM, CEM-DNR, K562 y K562-TAX), pulmón (A549) y células de osteosarcoma (U2OS). También se detectó una buena selectividad en los compuestos 11c y 11e para células cancerosas leucémicas y colorrectales (con y sin deleción de p53) (en comparación con MRC-5). Con respecto al compuesto 11a, exhibió una selectividad moderada (en comparación con las líneas celulares BJ y MRC-5) para las células A549; sin embargo, presentó buena selectividad (en comparación con las células BJ y MRC-5) para las células cancerosas derivadas de pulmón, carcinoma de colon, así como en células leucémicas y de osteosarcoma.
Como era de esperar, la citotoxicidad de los compuestos evaluados se observa significativamente afectada por el estado de oxidación del átomo de azufre unido a la posición C-4 de la quinolina y por la oxidación del nitrógeno quinolínico, como ha sido reportado previamente [49], y por el número y patrón de sustitución en que se encuentran los grupos metoxilos en el núcleo de la 1-indanona.
Análisis del ciclo y muerte celular
Por otro lado, queríamos averiguar si los compuestos 11a-e pueden detener el ciclo celular de las células cancerosas, como se reportó anteriormente para la cloroquina y análogos [49]- [54]. Con el fin de obtener una descripción más detallada de la actividad biológica de los derivados estudiados, realizamos un análisis del ciclo de las células CCRF-CEM más sensibles después de 24 h de incubación con los nuevos derivados de 7-cloro-(4-tioalquilquinolina) (Tabla 2).
Tabla 2 Citotoxicidad de los compuestos 11c-e en el ciclo celular en linfoblastos CCRF-CEM.
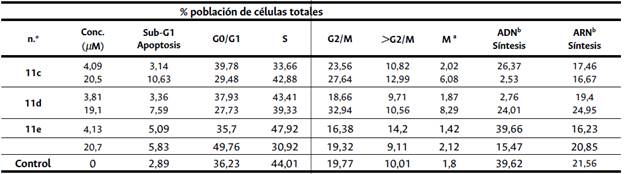
a Mitosis Fosfo-Histona3 (Ser10).
b Síntesis de ADN y ARN en linfoblastos CCRF-CEM (% de células positivas). Se usó análisis de citometría de flujo para cuantificar la distribución del ciclo celular y el porcentaje de células apoptóticas. La suma del porcentaje de G0/G1, S y G2/M es igual al 100%.
Se determinó el efecto de los compuestos en la distribución del ciclo celular para obtener información sobre el mecanismo de su actividad antiproliferativa. Como puede verse en la Tabla 2, una exposición de 24 h de células CCRF-CEM a concentraciones supreso-ras del crecimiento de derivados de 7-cloro-(4-tioalquilquinolina) (1 x CI50 y 5 x CI50 μM) resultó en una acumulación significativa de células en fase G2/M que se acompañó de un aumento de células con G0/G1 y una disminución del contenido de S, ADN y ARN. Todos los compuestos probados exhibieron un aumento dependiente de la dosis en la población de células mitóticas (pH3Ser10 positivas). Por ejemplo, en comparación con el control, el porcentaje de células en la fase G2/M aumentó mediante el tratamiento con 5 x CI50 /UM de compuestos durante 24 h (Tabla 2). Los compuestos también indujeron distintos valores sub-G1, que representan la población de células apoptóticas y muertas. Como se muestra en la Tabla 2, hubo un marcado aumento en el sub-G1 comparado con el valor de 2,89 en las células no tratadas. La 5-bromo-2-desoxiuridina (BrDU) se incorpora al ADN recién sintetizado y, por lo tanto, el marcado de pulsos de 5-bromouridina (BrU) se usa comúnmente como marcador de proliferación. La baja incorporación de BrDU y BrU en el ADN o el ARN, respectivamente, de las células tratadas con todos los compuestos a 5 x CI50 reflejó la inhibición de la síntesis de ADN y ARN, lo que indica cambios apoptóticos irreversibles. El porcentaje de células BrU negativas que incorporan 5-bromouridina es proporcional a la actividad transcripcional de las células CCRF-CEM.
Estos resultados sugirieron que estos compuestos podrían bloquear el ciclo celular e inducir la apoptosis y la muerte en las células CCRF-CEM de forma dependiente de la dosis in vitro.
Conclusiones
En este trabajo describimos un método conveniente para la síntesis de una mezcla de diasteroisómeros derivados de [(7-cloroqui-nolin-4-il)tio]alquil-1-indanones 11a-e a través de la reacción de [(7-cloroquinolin-4-il)tio]alcohol, previa oxidación con el reactivo de Dess-Martin para obtener el aldehído respectivo, que fue acoplado con las 1-indanonas correspondientes usando LDA. Los compuestos fueron evaluados por su potencial acción como citotóxicos con efecto in vitro a concentraciones micromolares contra las células derivadas del carcinoma de pulmón y colorrectal. Los compuestos, con excepción de 11b, muestran una buena selectividad sobre la proliferación de líneas celulares cancerosas con baja toxicidad para los fibroblastos MRC o BJ no malignos. Se observó una baja citotoxicidad frente a líneas celulares de cáncer multirresistentes (CEM-DNR, K562-TAX), lo que sugiere que son sustratos para transportadores farmacológicos. A concentraciones más altas (5 x CI50) de los compuestos 11c-e, contra la línea celular de cáncer CCRF-CEM, observamos la acumulación de las células en la fase celular G0/G1, la inhibición de la síntesis de ADN y ARN y la inducción de la apoptosis. El mecanismo molecular de este último efecto requiere investigación adicional.

