











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink1 Introducción
Se ha reconocido a lo largo de los años, que diferentes afecciones cardíacas están caracterizadas por la presencia de células incapaces de producir la energía necesaria para la contracción y relajación del miocardio, dicha deficiencia, ha sido vinculada con un mal funcionamiento mitocondrial. Las cardiopatías son enfermedades que representan un problema de salud pública en Colombia y en el mundo, debido al alto costo que representa su atención y tratamiento. En Colombia, se estima que el número de casos para dichas enfermedades van en aumento, a causa del ascenso en el envejecimiento de la población, y debido a diversos factores de riesgo cardiovascular, como es la predisposición genética y el estilo de vida 1.
Según el reporte generado por el Ministerio de salud y de protección social, para el año 2012, se registraron aproximadamente 1.1 millones de colombianos con falla cardíaca, con una prevalencia mayor en hombres (59.7 %) que en mujeres (40.3 %) 1. Las mitocondrias son organelas que participan en diversas vías biosintéticas y catabólicas, como: la oxidación de ácidos grasos, carbohidratos y proteínas, y los procesos de transporte de electrones y fosforilación oxidativa. Mediante dichos procesos, se genera la energía necesaria para suplir las demandas energéticas de los diferentes tejidos aerobios 2. Las células del músculo cardíaco participan en los procesos de contracción y relajación; proceso requerido para el bombeo de sangre oxigenada a los diferentes tejidos del cuerpo, es por tal razón, que dichas células presentan un gran número de mitocondrias, con el fin de suplir la gran demanda energética requerida para el buen funcionamiento del corazón 3. La actividad del miocardio se ve afectada, cuando diversas condiciones extracelulares e intracelulares como mutaciones, daño oxidativo, condiciones ambientales adversas y el envejecimiento, generan mitocondrias con grandes alteraciones estructurales y funcionales, lo que conlleva, a la activación de los procesos de control de calidad mitocondrial y de muerte celular (autofagia). Sin embargo, cuando dichos procesos no son efectuados de forma apropiada, ya sea, por condiciones de estrés crónico o deterioro por el envejecimiento, se genera la proliferación de mitocondrias disfuncionales en las células del corazón 4. Se ha identificado en pacientes con insuficiencia cardíaca, la presencia de mitocondrias cardíacas con disfunciones en la cadena transportadora de electrones, así como, en el proceso de síntesis de ATP (adenosín trifosfato; molécula energética) 5,6,7. Dicha disfunción mitocondrial, se ha relacionado con mutaciones en los genes codificantes de los complejos mitocondriales 8 o en genes que codifican para enzimas que participan en el proceso de β-oxidación de ácidos grasos; ruta catabólica que proporciona el 90 % de la energía requerida por el funcionamiento del corazón 9. Investigaciones realizadas en animales y humanos, han determinado que la acumulación de ciertos daños o mutaciones en el ADN mitocondrial a lo largo de la vida, llevan a un mal funcionamiento del sistema de fosforilación oxidativa, al aumento de especies reactivas de oxígeno (denomina ROS por su siglas en ingles) y a la aparición de cardiomiopatías 3. Debido a lo expuesto anteriormente, es de vital importancia conocer el papel de las mitocondrias en la disfunción cardíaca, con el fin de entender los mecanismos que participan en el correcto funcionamiento del corazón y cómo anomalías en estos procesos pueden llegar a relacionarse con cardiomiopatías.
Lo que se pretende con esta revisión, es recopilar el estado del arte actual encontrado mediante la búsqueda en base de datos virtuales como PUBMED, así como en libros de medicina y bioquímica, que permitan dar a entender el impacto de la función mitocondrial en las patologías cardíacas, debido a que estas van en aumento en la población Colombiana, y por ende, es trascendental aumentar el conocimiento en el tema, para establecer nuevos enfoques de estudio que se dirijan a la mitocondria como punto central para la creación de estrategias que regulen las alteraciones mitocondriales presentes en una determinada cardiomiopatía, de este modo, se generaran herramientas de prevención y tratamiento de las enfermedades cardíacas, contribuyendo a mejorar día a día la calidad de vida de los pacientes que padezcan una afección cardíaca determinada.
2 Función mitocondrial y su rol en el corazón
Las mitocondrias, son organelas intracelulares que se localizan en el citosol de la mayoría de células eucariotas, y su cantidad en cada célula dependerá de la función y demanda energética de cada órgano al que pertenecen dichas células; un ejemplo de ello, son los cardiomicitos; los cuales presentan alrededor del 38 % de su volumen en mitocondrias, debido a la alta demanda energética que requiere el corazón para su buen funcionamiento 2. Estas organelas, además de ser el punto central donde se produce la mayor cantidad de energía, por medio de la fosforilación oxidativa, también participan en diversos procesos biosintéticos (síntesis de proteínas, lípidos, síntesis de compuestos hemo, entre otros) y catabólicos (oxidación de ácidos grasos, cuerpos cetónicos y aminoácidos, ciclo de la urea, entre otros) 2. Las mitocondrias están compuestas por dos membranas; una membrana externa, que permite la importación y exportación de diversas moléculas desde el citosol a la mitocondria y viceversa, y una membrana interna, que se pliega en la matriz formando crestas que incrementan el área superficial. Entre las dos membranas mitocondriales, se encuentra el espacio intermembranal (IMM), el cual alberga al transportador móvil de electrones, citocromo C 10. Sobre la membrana interna, se encuentran proteínas integrales que representan los cuatro complejos (complejo I-IV) de la cadena respiratoria (cadena transportadora de electrones), el complejo ATP sintasa (F1F0-ATPasa) y el transportador de nucleótidos de adenina (ANT), así como, diversos trasportadores de sustratos como el piruvato. El transporte de electrones, a través de cada uno de los cuatro complejos, genera el gradiente eléctrico necesario para la síntesis de ATP. Este gradiente, se define como ∆µH+, el cual, incluye al potencial de membrana (∆Ψ) y al gradiente protónico (∆pH) 10. En la matriz mitocondrial, está localizada la maquinaria necesaria para la producción de 13 proteínas de la cadena respiratoria (Tabla 1). Estas proteínas están codificadas en el material genético propio de la mitocondria; el cual es una molécula de ADN circular de poco tamaño al compararlo con el ADN nuclear de las células. Por ejemplo, una célula humana diploide (2n) tiene en su núcleo, aproximadamente, seis mil millones (∼6000,000,000) de pares de bases nitrogenadas, organizadas en 46 cromosomas (paquetes lineales independientes), mientras que su mitocondria tiene moléculas de ∼ 16,550 pares de bases, en múltiples copias. La integridad del material genético mitocondrial determinará la apropiada formación de los complejos necesarios para el transporte de electrones y por lo tanto, el rendimiento energético de las células 11.
Tabla 1 Genes humanos codificados por el ADN mitocondrial organizado de acuerdo a su participación en los complejos mitocondriales. Tomada de Stewart y Chinnery [12]

En la mayoría de las membranas biológicas, la relación entre proteínas y lípidos es alrededor del 50:50, respectivamente, sin embargo; la membrana interna mitocondrial se caracteriza por presentar una proporción de aproximadamente, 75:25 de proteína y lípidos, respectivamente; siendo el fosfolípido cardiolipina, el lípido característico de la membrana interna mi tocondrial y el cual contribuye, con el buen funcionamiento de las proteínas mitocondriales (2). El corazón principalmente bombea sangre oxigenada y nutriente a todos los tejidos del cuerpo, para este propósito, se generan continuamente contracciones musculares, las cuales son inducidas por la liberación de Ca2+ desde el retículo sarcoplásmico de los cardiomiocitos, al citoplasma de las mismas (12). Los iones Ca2+, ingresan a las mitocondrias y regulan las enzimas del ciclo del ácido tricarboxílico (isocitrato deshidrogenasa, ?-cetoglutarato deshidrogenasa y la malato deshidrogenasa), lo que permite la descarboxilación del acetil-CoA y el catabolismo de carbohidratos, aminoácidos, y principalmente, la β-oxidación de ácidos grasos, para generar equivalentes reductores, como el NADH y el FADH2, los cuales, son oxidados por la cadena transportadora de electrones (Figura 1). En dicho proceso, se genera una acumulación de protones en el espacio intermembranal (IMM), activando de forma indirecta la ATP sintasa, la cual bombea nuevamente los protones hacia la matriz mitocondrial, para estabilizar el gradiente electroquímico y producir ATP (12). Diversas alteraciones intracelulares, como mutaciones genéticas, o alteraciones extracelulares, como factores ambientales, generan un aumento en la producción de radicales libres, así como daños mitocondriales. Para la restauración de las condiciones normales, las mitocondrias poseen diferentes mecanismos que permiten el control de su calidad; entre estos se encuentran la fisión y la fusión mitocondrial. La fisión, permite generar a partir de una mitocondria, dos mitocondrias pequeñas; este proceso es promovido por las proteínas relacionadas con la dinámica (DRP1), proteína 1 de la fisión mitocondrial (FIS1) y factor de fisión mitocondrial (MFF). Por medio de la fisión, se aumenta la resistencia al estrés oxidativo y se disminuye la segregación de mitocondrias disfuncionales con daños irreversibles mediante su eliminación por mitofagia (autofagia) (13). La mitofagia, es el mecanismo mediante el cual las mitocondrias dañadas son incorporadas en un autofagosoma para ser posteriormente degradadas por los lisosomas (13). El proceso de mitofagia en los mamíferos es promovido y regulado por: la quinasa 1 (PINK1); localizada en la membrana externa mitocondrial, y una ligasa ubiquitina E3 (Parkin); localizada en el citosol. En presencia de estrés celular, Parkin es estimulada por PINK1 para ser translocada desde el citosol hasta las mitocondrias dañadas, con el fin, de dirigir la fragmentación mitocondrial e iniciar la mitofagia (14),(15). Las proteínas PINK1 y Parkin se han relacionado con la prevención de la muerte celular, mediante el mantenimiento de la función mitocondrial (14). Por otro lado, la fusión mitocondrial, permite la unión entre la membrana interna y externa de dos mitocondrias para generar una sola mitocondria alargada. Mediante este proceso, se generan mitocondrias con gran capacidad oxidativa, se permite la reparación de daños reversibles y se limita la aparición de ADN mitocondrial mutado durante el envejecimiento. Este mecanismo, está controlado por las proteínas mitofusinas 1 y 2 (MFN1 y MFN2) y la atrofia óptica 1 (OPA1) (13). Los procesos de fusión, fisión y mitofagia mitocondrial son necesarios para regular la homeostasis cardíaca y la adaptación al estrés. Investigaciones en ratones han determinado que deleciones en los genes MFN2 y OPA1 de células cardíacas, generan el desarrollo de trastornos cardíacos como la hipertrofia ventricular. Igualmente, se ha determinado que la supresión del gen DRP1 (fusión) se asoció con el desarrollo de dilatación ventricular (13).
En muestras de corazón humanos con falla cardíaca, se ha evidenciado una disminución en los niveles de OPA1, por lo que dicha proteína, se ha asociado a la preservación de la estructura de la membrana interna mitocondrial y a la protección celular frente a la apoptosis (16). Acorde a lo anterior, en el estudio de Billia et al. (17), realizado en ratones con deleción en el gen PINK1, se demostró un aumento en el estrés oxidativo y daño mitocondrial, lo que conllevo, al desarrollo de disfunción del ventrículo izquierdo e hipertrofia cardíaca. Los autores reconocieron que los niveles de PINK1 son cada vez menores en mitocondrias de corazón humano con falla cardíaca.
Por tanto, la pérdida de los mecanismos de control mitocondrial junto con la inhibición de la mitofagia ocasionada por el envejecimiento, estrés hemodinámico o diversos inductores externos, conllevan, al aumento del estrés oxidativo y a la acumulación de mitocondrias disfuncionales, las cuales, presentan una serie de anormalidades funcionales y estructurales, que disminuyen la eficiencia del proceso de respiración celular y por ende, la síntesis de ATP, influyendo así; en la generación de diferentes afecciones cardíacas (13).
3 Subpoblaciones mitocondriales cardíacas
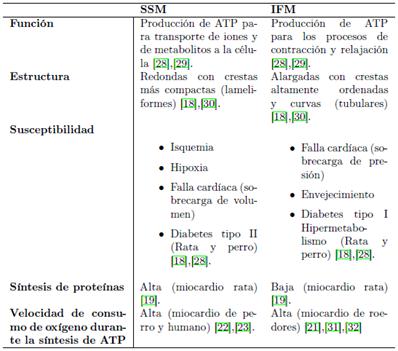
Las mitocondrias presentes en los cardiomiocitos de diversos mamíferos se han estudiado por microscopía electrónica de transmisión, lo que permitió evidenciar la presencia de mitocondrias elípticas, con numerosas crestas transversales; las cuales son laminiformes o tubulares. Así mismo, por medio de dicha microscopia, lograron identificar la presencia de dos subpoblaciones mitocondriales dentro de los cardiomiocitos: las denominadas (1) subsarcolemmal mitochondria (SSM), que están localizadas debajo del sarcolema y presentan una forma intracelular redonda, con baja densidad electrónica en la matriz y crestas laminiformes (Figura 1), y las (2) interfibrillar mitochondria (IFM) que se encuentran distribuidas entre las miofibrillas de actina-miosina y presentan una forma alargada, con alta densidad electrónica y crestas tubulares (18) (Figura 1).

Figura 1: Micrografía electrónica de la localización y morfología de mitocondrias SSM e IFM de miocardio de rata. A. Localización de las subpoblaciones mitocondriales dentro de las células cardíacas. B. Morfología de las subpoblaciones mitocondriales después de su aislamiento. Tomada de Holmuhamedov et al. (18)
Diversas investigaciones han determinado que estas dos subpoblaciones difieren a nivel funcional, bioquímico, y en la composición proteica y lipídica, así como, en la síntesis de proteínas (Tabla 1). Kasumov et al. (19), mediante el análisis de tejido proveniente del ventrículo izquierdo de ratas sanas, determinaron que la velocidad de síntesis de proteínas en SSM era mayor en un 15 %, con respecto, a la síntesis de proteínas en IFM. Se ha determinado igualmente, que ambas subpoblaciones mitocondriales, difieren en la sensibilidad que presentan frente a cambios metabólicos, o frente a diferentes patologías (18),(20).
En la investigación realizada por Palmer et al. (21) hace aproximadamente 40 años, encontraron diferencias bioquímicas entre ambas subpoblaciones mitocondriales, observando que las IFM presentaron una velocidad de consumo de oxígeno para el estado 31 superior en un 44 %, en comparación con la velocidad de consumo de oxígeno determinada para las SSM. Por el contrario, Rosca et al. (22) y Croston et al. (23), reportan velocidades de consumo de oxígeno para el estado 31, superiores en SSM en comparación a las obtenidas para IFM en mitocondrias de miocardio de perro y humano, respectivamente. Aunque varias investigaciones han evidenciado la existencia de diferencias entre las dos subpoblaciones mitocondriales cardíacas, hasta la fecha, no hay estudios que especifiquen con claridad la causa de dichas diferencias, sin embargo, en diferentes publicaciones se han implicado a factores como: las concentraciones de fosforo inorgánico (25), concentración de Ca2+ (18), la velocidad de síntesis de proteínas (19), el perfil lipídico de la membrana interna mitocondrial (26), y a la morfología de la membrana interna mitocondrial2 (27), en las variaciones bioquímicas existentes en el sistema de fosforilación oxidativa entre las mitocondrias SSM e IFM.
4 Disfunción cardíaca asociada a mitocondrias disfuncionales
Numerosas investigaciones, han reconocido que la disfunción mitocondrial es uno de los factores determinantes en las alteraciones cardíacas, como: la falla cardíaca, la isquemia/reperfusión y el envejecimiento 10,33. La pérdida de la función mitocondrial origina un incremento en la producción de ROS y alteraciones en el sistema de fosforilación oxidativa, lo que conlleva, al aumento de la muerte de los cardiomiocitos a través de la liberación de factores apoptogénicos3. Por ende, la pérdida de la función mitocondrial, así como, de cardiomiocitos genera una reducción en la producción de la energía necesaria para el funcionamiento del corazón 30. El Observatorio Nacional de Salud Colombiano 34 ha determinado que las enfermedades cardiovasculares fueron una de las principales causantes de muerte en Colombia en el periodo entre 1998 y 2011. Estas enfermedades, representaron el 23.5 % del total de las muertes en el país, dentro de las cuales el 56.3 % fueron por enfermedad cardíaca isquémica, el 30.6 % por enfermedad cerebrovascular, el 12.4 % por enfermedad hipertensiva y el 0.5 % por enfermedad cardíaca reumática crónica.
Se ha reconocido, que condiciones como el envejecimiento, la diabetes tipo II y la obesidad están relacionadas con el aumento de patologías cardíacas como el infarto de miocardio; el cual se ha ido convirtiendo en uno de los principales problemas clínicos a nivel mundial 33. El envejecimiento, es considerado una de las condiciones que contribuye con la aparición de cardiomiopatías, debido a que en dicha condición, han observado una disminución en la actividad del complejo III y IV, así como, una reducción en la reserva bioenergética y un aumento del estrés oxidativo en mitocondrias de diversos roedores y primates de avanzada edad 35. Frente a las alteraciones mitocondriales, se reconoce que niveles bajos de la actividad enzimática de los complejos I, III y IV, se relacionan con alteraciones de la cadena transportadora de electrones de mitocondrias cardíacas de modelos animales o humanos con hipertrofia y miocardiopatía 5,6. Griffiths et al., 7, lograron identificar en conejos neonatos con hipertrofia inducida, alteraciones progresivas en la cadena transportadora de electrones, por deterioro de los complejos mitocondriales I y II, dicha alteración, contribuyo a una diminución en la producción de ATP. Igualmente, los investigadores observaron baja producción de ROS (peróxido de hidrogeno) y un aumento en la perdida de cardiomiocitos por apoptosis. Así mismo, en ratas con infarto de miocardio Heather, et al. 36 evidenciaron un aumento de ROS, debido a una disminución en la actividad del complejo III, lo que estuvo acompañado de defectos en el transporte de electrones desde el complejo III al complejo IV. Para lo anterior, los autores conjeturaron que dicha deficiencia en el complejo III se asoció con un deterioro en la proteína hierro-azufre. Andreu et al. 8 en pacientes con cardiomiopatía histiocitoide identificaron células cardíacas de tipo histocito granulares y espumosas, así como, mutaciones en uno de los nucleótidos presentes en el ADN mitocondrial, asociado al gen que codifica para la subunidad cty b del complejo III. Dicha mutación, generó defectos mitocondriales relacionados con fallas en la cadena de transportadora de electrones. Con relación a lo anterior, defectos o alteraciones en las subunidades del complejo III mitocondrial generan fallas en el sistema de fosforilación oxidativa 2, ya que mediante el ciclo Q se da la transferencia de electrones desde el complejo I y II al citocromo C. Bloqueos en dicha vía, promueven la fuga de electrones y la disminución del gradiente electroquímico en el IMM, lo que ocasiona una reducción en la síntesis de ATP 36. Es por ello, que la estabilidad del complejo III es esencial para mantener la bioenergética del miocardio. De igual forma, se ha determinado que niveles bajos de coenzima Q, están presentes en enfermedades cardíacas, como: insuficiencia cardíaca congestiva, cardiopatía isquémica y angina de pecho. Lo anterior, se debe a que cambios en los niveles de coenzima Q, están asociados con alteraciones en la cadena de transporte de electrones, ya que dicho transportador móvil, es esencial para el transporte de los electrones desde el complejo I y II hasta el complejo III mitocondrial, además de ello, éste es considerado un captador de radicales libres y un protector celular 37.
4.1 Disfunción de las subpoblaciones mitocondriales cardíacas (subsarcolema (SSM) e Inferftbrilares (IFM))
Como se mencionó en la Tabla 2, las subpoblaciones mitocondriales presentan diferencias disfuncionales frente a diferentes patologías. En múltiples investigaciones se ha reconocido que la insuficiencia cardíaca, la diabetes, el envejecimiento, y la hipertensión están asociadas con alteraciones en las dos subpoblaciones mitocondriales 19. En miocardio con insuficiencia cardíaca, se ha determinado que las IFM presentan defectos en los procesos de fosforilación oxidativa, contrario a las SSM las cuales permanecieron intactas 9. Rosca et al. 6, investigaron las mitocondrias SSM e IFM obtenidas de corazón de perros con miocardiopatía dilatada idiopática (IDC). En el estudio, se observó que las SSM aisladas del ventrículo izquierdo no presentaron una disminución en la actividad del complejo I, II y III, en comparación con las IFM; las cuales presentaron una disminución en la actividad de dichos complejos. A partir de lo hallado, los autores sugirieron la existencia de alteraciones en las mitocondrias interfibrilares por presencia de defectos en los componentes y organización de las proteínas mitocondriales. Para lo anterior, los investigadores asociaron sus resultados con los encontrados en diversos experimentos realizados en hámsters con miocardiopatía, donde se evidencio alteraciones en el proceso de fosforilación oxidativa debido a una disminución en el estado respiratorio 3 de mitocondrias IFM. Contrario a lo anterior, Ruiz-Meana, et al. 38, aislaron mitocondrias de corazón de ratas con isquemia/reperfusión inducida y reconocieron una reducción en la velocidad de consumo de oxígeno, con una baja actividad del complejo I y II, así como, un aumento en la producción de superóxido en SSM, mientras que las IFM, no presentaron cambios en velocidad de consumo de oxígeno. Los autores, lograron atenuar las alteraciones presentes en las SSM mediante el precondicionamiento isquémico4, para lo cual determinaron que la proteína Conexima 43 (proteína de membrana) puede llegar a estar involucrada en el proceso de precondicionamiento isquémico debido a que esta se encontró principalmente en SSM.
A nivel fisiológico, se ha observado que las enfermedades cardiovasculares se caracterizan, por: cambios morfológicos en las células cardíacas, cambios bioquímicos en los procesos de señalización y en las propiedades eletromecánicas, así como, cambios en los componentes de la matriz extracelular, con un subsecuente aumento de la rigidez del tejido cardíaco. Eventualmente, debido a dichas alteraciones se da paso a la muerte de los cardiomiocitos y a la aparición de insuficiencia cardíaca 39. Acorde con lo anterior, Dague et al. 39, usando microscopía de fuerza atómica (AFM), encontraron alteraciones estructurales en mitocondrias SSM y en el sarcolema de cardiomiocitos de corazón de rata con falla cardíaca. Dichas alteraciones estructurales, se relacionaron con desorganización del sarcolema y pérdida de la topología de las crestas mitocondrias de SSM, mientras que las IFM, se encontraron alineadas entre los miofilamentos, sin alteración alguna en la etapa temprana de la insuficiencia cardíaca. En la etapa terminal de la insuficiencia cardíaca post-isquemica, hubo aumento de la rigidez de la superficie celular, con agotamiento de las SSM, junto con pérdida de los túbulos T (Invaginaciones del sarcolema), así como, un desplazamiento de las IFM hacia la membrana. Los investigadores sugirieron, que un determinado estrés inicial en el sarcolema, genera la pérdida de SSM, junto con el desarreglo de los túbulos T y la aparición de la insuficiencia cardíaca. Como ya se ha mencionado, el envejecimiento causa pérdida de la función mitocondrial, lo que conlleva a un aumento en la muerte de los cardiomiocitos a través de la liberación de factores apoptogénicos; proceso mediado por dichas mitocondrias disfuncionales. En el envejecimiento se presentan alteraciones bioquímicas en IFM; caracterizadas por una disminución en el rendimiento proteico, así como, en la velocidad de fosforilación oxidativa, mientras que las SSM, no presentan dichas alteraciones 30. Sin embargo, en el estudio realizado por Hofer et al. 40 en mitocondrias SSM e IFM aisladas de corazón de rata, determinaron que con la edad no hubo aumento en la generación de H2O2, ni disminución en la velocidad de consumo de oxígeno en IFM y SSM. No obstante, las IFM se mostraron más susceptibles a estrés por Ca2+ (pérdida de capacidad de retención de calcio), susceptibilidad que es evidenciada en la isquemia/reperfusión, la cual involucra fuertes contracciones. En el envejecimiento, también se ha observado, que las células cardíacas pierden su capacidad de utilizar ácidos grasos como sustrato. Gómez et al. 35, encontraron en miocardio de rata entre 24 a 28 meses, una disminución del 28 % en la actividad de la carnitina palmitoiltransferasa 1 (CPT1) de mitocondrias IFM, así mismo, para este tipo de mitocondrias también encontraron una disminución en el estado respiratorio 3, al usar como sustrato el palmitoyl-CoA. Por el contrario, las SSM preservaron la actividad de CPT1 y utilizaron eficiente el palmitato como sustrato. Schwarzer et al. 32, evaluaron las SSM e IFM de corazón de rata macho que presentaron hipertrofia ventricular inducida, en el estudio, los autores encontraron una reducción en el estado respiratorio 3 para las IFM, junto con la aparición de disfunción contráctil en el corazón. Con base a las evidencias obtenidas en el estudio, se soporta la idea de que las IFM producen el ATP principalmente para la función contráctil. En soporte a lo anterior, Kadambari et al. 9, sugirieron que las IFM contribuyen a la disfunción contráctil en ratas con insuficiencia cardíaca, debido a que encontraron una ineficiente producción de ATP, la cual, fue asociada con una reducción en velocidad de consumo de oxígeno para el estado 3, al usar como sustrato palmitoil-carnitina. Por ende, se indicó una disminución en la capacidad de oxidación de dicho sustrato, junto con una disminución de la actividad de CPT1 y de los complejos mitocondriales.
4.2 Generación de energía en mitocondrias disfuncionales del músculo cardíaco
La producción de moléculas de ATP es generada en menor proporción, en las células del músculo cardíaco que presentan mitocondrias disfuncionales, lo que contribuye, a alteraciones en el metabolismo energético aerobio del corazón de personas que padecen diversas afecciones cardíacas. En la insuficiencia cardíaca, existe un desequilibrio entre la función que el corazón debe realizar y la energía que este es capaz de producir para realizar su trabajo. Dicha deficiencia en la producción de energía se debe a limitaciones en el sistema de fosforilación oxidativa y a una disminución en la oxidación de ácidos grasos; proceso mediante el cual se obtiene el 90 % de la energía requerida para el funcionamiento del corazón 4. En investigaciones experimentales, se ha determinado que las enfermedades cardíacas presentan un decrecimiento de la actividad del complejo ATP sintasa, representada por una disminución del 14 % en la relación ADP/O5 3. En estudios realizados en mitocondrias de corazón de cerdo con insuficiencia cardíaca isquémica, así como, en perros y humanos con miocardiopatía dilatada, se encontró disminución en la cantidad y actividad de la ATP sintasa 6.
Rosca et al. 5, encontraron en mitocondrias aisladas de corazón de perro con insuficiencia cardíaca inducida una disminución de la actividad del complejo ATP sintasa. Las IFM no presentaron fallas en la cadena transportadora de electrones, pero sí, en el proceso de fosforilación oxidativa a nivel de la ATP sintasa y de los transportadores de ADP y ATP (isoformas ANT1 y ANT2), para los cuales, se encontró una mayor proporción del transportador ANT2. En las SSM, evidenciaron alteraciones en el proceso de fosforilación oxidativa, ocasionada por daños en la cadena transportadora de electrones, además, de encontrar una mayor proporción del transportador ANT2. Para esta investigación, se determinó que cambios en la isoforma de los transportadores de ADP/ATP, ocasionan cambios en las propiedades cinéticas; donde la ANT2 presenta mayor eficiencia en el transporte, en comparación con la ANT1. Debido a lo anterior, se puede deducir que las mitocondrias disfuncionales, no sólo presentan daños en su mecanismo de transporte de electrones por medio del cual se proporciona la energía necesaria para inducir la síntesis de ATP, sino también, en los transportadores de los sustratos requeridos para la producción de equivalentes reductores y de ATP, lo que genera una reducción en la producción de la energía requerida para el funcionamiento del corazón.
5 Disfunción mitocondrial originada por infecciones e inflamaciones cardíacas
Diversos factores ambientales, como compuestos contaminantes o tóxicos, así como, la presencia de virus, bacterias, hongos y parásitos en el ambiente, contribuyen con la generación de infecciones cardíacas caracterizadas por la inflamación del corazón 42. Dichas inflamaciones cardíacas se dividen en tres tipos: miocarditis, periocarditis y endocaditis (Tabla 3).
Diferentes procesos infecciosos generan alteraciones mitocondriales, mediante la manifestación de procesos inmunológicos, como la producción de citocinas y óxido nítrico, que combaten la acción de diversos patógenos, pero también, generan disfunciones mediante la producción de inflamación y estrés oxidativo. En mitocondrias cardíacas de modelos animales como la rata, se ha observado en respuesta a la inflamación cardíaca, un aumento en la oxidación de lípidos y proteínas mitocondriales, alteraciones en los mecanismos de defensa contra las ROS, daño en el perfil lipídico de la membrana mitocondrial y en la integridad de esta. Lo anterior, es asociado con alteraciones de la cadena transportadora de electrones, y con la liberación de citocromo C, el cual induce a la apoptosis celular y a la aparición de cardiomiopatías 43.

Dentro de las patologías inflamatorias, se ha encontrado en la enfermedad de Chagas, es una de las causas de la miocarditis crónica. Chagas se origina por la infección con el parasito Trypanosoma cruzi, dicho parasito tiene la capacidad de reproducirse dentro de los cardiomiocitos, lo que conlleva, a un aumento del estrés oxidativo, afectando principalmente la estructural y funcionalidad de las mitocondrias cardíacas 58, y generando dilatación ventricular, arritmia y disfunción contráctil 58. Wen et al. 46, estudiaron la bioenergética mitocondrial a partir de aislados obtenidos de ratones infectados con Trypanosoma cruzi. En el estudio, los autores observaron una reducción significativa en la actividad del complejo III debido a deleciones en la subunidad cty b. Dicha mutación en el complejo III, generaron fallas en el funcionamiento del sistema de fosforilación oxidativa; caracterizado por una disminución de la velocidad respiratoria del estado 3, y en la producción de ATP. Disminuciones en los niveles de energía, también se han observado en el corazón de modelos humanos 47, por lo cual se asume que las alteraciones mitocondriales contribuyen con la disfunción contráctil del miocardio 46.
Acorde con lo anterior, Wen et al. 59, evaluaron la acción del antioxidante α-fenil-N-tert-butilnitrona y el antiparasitario benzonidazol para el trato de ratas con la enfermedad de Chagas. En el estudio, lograron identificar que la combinación de ambos compuestos redujo las reacciones inflamatorias, así como los niveles de ROS, preservando así el funcionamiento de la cadena respiratoria y la producción de energía en el corazón.
6 Implicación de los supercomplejos en la disfunción cardíaca
Por algún tiempo, se estableció que el proceso de transporte de electrones estaba formado a partir de una organización aleatoria de los complejos mitocondriales individuales (Modelo de difusión aleatoria) entre la membrana interna mitocondrial. A partir de este modelo, el transporte de electrones ocurría gracias a transportadores móviles como la coenzima Q y el citocromo C, los cuales se difunden libremente por la membrana lipídica. Aun así, desde el año 2000 se ha venido discutiendo sobre la organización de los complejos mitocondriales en forma de asociaciones como supercomplejos; se ha determinado las asociaciones entre los complejos I1III1IV1 o I1III2 IV1, principalmente en mamíferos 60,61. En mitocondrias de corazón de bovino, se ha determinado la presencia del complejo I1III2 con un 17 % del contenido total del complejo I y el supercomplejo I1III2IV1 y I1III2IV2 los cuales contenían el 54 % y el 9 % del total del complejo I, respectivamente 60,62. La estabilidad del complejo I se ha correlacionado con la estabilidad de los supercomplejos, frente a esto, se ha podido determinar que ciertas mutaciones en las subunidades del complejo III o IV se relacionan con la desestabilización del complejo I y la pérdida del supercomplejo I1III1IV1 en mitocondrias de ratón y humano. [62]. Así mismo, se ha estimado que un aumento en la producción de ROS contribuye a la desestabilización de los supercomplejos; mediante fallas en el ensamble del complejo I, lo cual, ha sido relacionado también con una reducción en la captación de electrones y en la síntesis de ATP 63. Las interacciones proteína-lípido se han denominado como factores claves para la estabilidad de los supercomplejos, un ejemplo de ello, es el aumento del estrés oxidativo que ocasiona daños en la membrana lipídica y en las proteínas mitocondriales, lo que contribuye con la desestabilización de los supercomplejos. Igualmente, se ha evidenciado que el fosfolípido cardiolipina, el cual se encuentra en la membrana interna mitocondrial permite las interacciones proteína-proteína. En estudios realizados sobre este fosfolípido han creado la hipótesis, de que alteraciones en éste, pueden generar desestabilización de los supercomplejos 35. En levaduras mutadas, han demostrado una correlación directa entre los niveles de cardiolipina y la formación de supercomplejos mitocondriales 64. Los supercomplejos han sido catalogados como esenciales para el funcionamiento eficiente de la cadena respiratoria, al promover el acoplamiento entre los complejos, la estabilidad de estos y una mayor eficiencia en la captación de sustratos y electrones, debido a que los transportadores móviles como la coenzima Q y el citocromo C permanezcan cerca a los complejos. La formación de supercomplejos, también supone, la reducción de fugas electrónicas, y por ende, la reducción de ROS. Es por lo anterior, que los supercomplejos se asocian a la regulación de la bioenergética mitocondrial 35,65. Sin embargo, Blaza et al. 66, descartan el papel cinético de los supercomplejos dentro de los procesos catalíticos, ya que argumentan que los transportadores móviles (coenzima Q y el citocromo C) se intercambian libremente en la estructura de los supercomplejos I-III2-IV, y por ende, para los autores los supercomplejos simplemente son interacciones débiles que permiten mantener concentraciones altas de proteínas mitocondriales. No obstante, se ha considerado que la desestabilización de los supercomplejos mitocondriales genera pérdidas en la bioenergética mitocondrial y dicha alteración está correlacionada con enfermedades como el Parkinson, el Alzheimer, el cáncer, el síndrome de Barth, la disfunción cardíaca y el envejecimiento 60. Igualmente, en mitocondrias de corazón de perro con insuficiencia cardíaca, han determinado deficiencia en el proceso de fosforilación oxidativa sin cambios en los niveles de los complejos mitocondriales individuales, por ello, se ha concluido que dicho déficit se puede deber a alteraciones en el ensamble de los complejos en forma de supercomplejos 6. Rosca et al. 65, realizaron sus estudios a partir de mitocondrias cardíacas del subsarcolema (SSM) e interfibrilares (IFM) obtenidas del corazón de perro con insuficiencia cardíaca. En sus experimentos, los autores identificaron una disminución del supercomplejo I1III2IV1 y un aumento en la cantidad de los complejos individuales I, III y IV, en ambos tipos de mitocondrias, así como, una reducción del 50 % en el proceso de fosforilación oxidativa. De la misma manera, Gómez et al. 67, investigaron en mitocondrias de corazón de ratas viejas la disociación de los supercomplejos mitocondriales, determinando que la concentración del supercomplejo I1III2IV1 se redujo de un 13 a un 25 % con la edad. Por consiguiente, los autores concluyeron que el envejecimiento se asocia a la reducción de los niveles de supercomplejos mitocondriales, pero dicha alteración no afecta los niveles de la mayoría de los complejos individuales de la cadena transportadora de electrones. Paralelo con lo anterior, y en pro de entender como está organizado el sistema de transporte de electrones Beutner et al. 68, al estudiar las mitocondrias obtenidas de corazón de ratones en estado embrionario, determinaron que el modelo de difusión aleatoria y el modelo de estado sólido eran transitorios. Los autores suponen, que frente a diferentes condiciones como el desarrollo celular, la cadena de transporte de electrones puede hacer una transición del estado de difusión aleatoria al estado sólido, para aumentar la eficiencia en la transferencia de electrones. Así mismo, Guaras et al. 69, propone la existencia de diferentes configuraciones organizacionales de la cadena transportadora de electrones para el control del flujo de los electrones provenientes del NADH y FADH2. Para dicha hipótesis, los investigadores plantean que la coenzima Q actúa como un sensor metabólico, controlando la producción de ROS y la actividad de la NADH deshidrogenasa, con el fin de activar el transporte de electrones inverso, en el cual el complejo III se desensambla del complejo I degradado, y se activa la función del complejo II con el objetivo de optimizando la capacidad oxidativa.
7 Rol de la cardiolipina en las mitocondrias
Durante varios años, se ha planteado la hipótesis de que los supercomplejos están estructuralmente ensamblados por la cardiolipina; fosfolípido presente en la membrana interna mitocondrial, y su forma predominante es la cardiolipina tetralinoleica ((18:2) 4CL). En varias investigaciones, se ha reconocido a éste fosfolípido como esencial para el buen funcionamiento de los complejos mitocondriales, y por tanto, se ha asociado con el transporte eficiente de electrones 65,70,71. Se presume, que en ausencia de la cardiolipina, los supercomplejos III1IV1 se desestabilizan, generando una disminución en la actividad de la cadena transportadora de electrones, del complejo IV y una reducción en el potencial de membrana. Por consiguiente, se ha sugerido que la cardiolipina está implicada en el buen funcionamiento del sistema de fosforilación oxidativa, así como, en el ensamble de los supercomplejos 71. Hasta el momento, no se conoce con precisión si la cardiolipina es esencial para la formación de supercomplejos, aunque en varios estudios, se ha dejado claro, que este fosfolípido se requiere para el correcto funcionamiento de los diferentes componentes de la cadena transportadora de electrones 63,65. Acorde con esto, la cardiolipina junto con la fosfatidiletanolamina ha sido implicada en la interacción proteína-proteína entre el complejo IV y el complejo III de levaduras 72. Del mismo modo, Peyta et al. 73, encontraron en células de hepatocitos humanos con mutaciones en el gen cardiolipina sintasa, una disminución del 45 % en el contenido de cardiolipina, el cual, se correlaciono con la desestabilización de los supercomplejos mitocondriales y con una reducción en el consumo de oxígeno (baja actividad de la cadena transportadora de electrones). Pokorná et al. 74, argumentan que un desequilibrio en relación con los fosfolípidos aniónicos de la membrana mitocondria, se relaciona con defectos en la morfología y función mitocondrial. Niveles bajos de cardiolipina en la membrana interna mitocondrial, están implicados en patologías como el síndrome de Barth. Dicho síndrome se relaciona con anomalías en la expresión del gen taffazin, ubicado en el cromosoma X. Alteraciones en el gen taffazin genera la producción de cardiolipinas inmaduras con variaciones en sus cadenas laterales acíl. Este síndrome, está correlacionado con cardiomiopatías, miopatía esquelética y retraso en el desarrollo de infantil 75,76. De igual forma, se ha demostrado que los niveles de cardiolipina, están afectados en pacientes que presentan miocardiopatía dilatada idiopática (IDC); la cual se correlaciona con daño en el proceso de contracción de los ventrículos, lo que genera, disfunción en la dilatación del músculo cardíaco. Chatfield et al. 76, determinaron que en niños y adultos con IDC se presentó un bajo contenido de cardiolipina tetralinoleoica. Igualmente, Petrosillo et al. 70, evidenciaron en mitocondrias de corazón de ratas con isquemia cardíaca, un aumento del estrés oxidativo que conllevo a la peroxidación de lípidos, junto con una disminución de los niveles de cardiolipina, y por ende, la perdida de actividad de los complejos I y III con disminución en la velocidad de consumo de oxígeno para el estado 3 y en la razón de control respiratorio.
8 Tratamientos enfocados en la recuperación de la función mitocondrial
Como se ha discutido a lo largo de la revisión, el correcto funcionamiento del corazón, depende en esencia, de un correcto funcionamiento mitocondrial; y por tanto, mutaciones o disfunciones en esta organela, desencadena en cardiopatías. Por consiguiente, desde hace algún tiempo, diversas investigaciones se han enfocado en la mitocondria como objetivo para el tratamiento de disfunciones cardíacas. Algunos de los posibles fármacos investigados para el tratamiento de la disfunción cardíaca se encuentran en la Tabla 4. Los fármacos se dirigen especialmente, hacia la optimización de la expresión de diferentes proteínas o cofactores como: el factor de transcripción mitocondrial A (TFAM), la proteína quinasa activada por adenosina monofosfato (AMPK) y la proteína 1α activadora del receptor activado por el proliferador de peroxisomas (PGC1α) 77. Dichas proteínas, intervienen en los procesos de transcripción y replicación del ADN mitocondrial 78, en la activación de proteínas mitocondriales de origen nuclear 79 y en la biogénesis mitocondrial 80, respectivamente. Por consiguiente, los tratamientos pretenden mejorar la función mitocondrial mediante la restauración funcional de los complejos mitocondriales 81, la potencialización frente la captación de diferentes iones como el Ca2+ y estabilización de la membrana plasmática, y de fosfolípidos como la cardiolipina, que intervienen en la bioenergética mitocondrial 83,84. Los fármacos evaluados a la fecha han sido considerados como cardioprotectores, ya que pretenden reducir los efectos adversos que impiden el correcto funcionamiento del corazón. En pacientes con falla cardíaca se ha evaluado el Resveratrol, el cual se encargó de estabilizar la función diastólica y endotelial 85, así mismo, la patente WO 2009108999 A1 indica la formulación de Resveratrol para la prevención o el tratamiento de patologías asociadas con estrés oxidativo y daño en el ADN 86. La CoQ10 en pacientes con falla cardíaca disminuyo los efectos cardiovasculares y protegió al miocardio contra la isquemia 83. La patente US 6331532 B1 hace referencia al uso antioxidantes cationicos lipofílicos como el mitoquinol para prevenir el daño mitocondrial por estrés oxidativo, así como, en la reducción de lesiones por isquemia/reperfusión o infarto 87.
Tabla 4: Tratamientos dirigidos a la mitocondria para el control de la disfunción cardíaca

*Poro de permeabilidad transitorio mitocondrial
La Deferiprona redujo la hipertrofia cardíaca en pacientes con ataxia de Friedreich, al reducir el daño oxidativo generado por los altos niveles de hierro en mitocondrias 88,89. La patente US 20130190365 A1 establece las cantidades adecuadas de dicho fármaco para la prevención, estabilización y tratamiento de la ataxia de Friedreich 90. En cuanto al uso de CGP-37157, la patente US 20120077763 A1 especifica el uso de dicho fármaco para controlar los efectos tóxicos de los glucósidos, así como, para la prevención y control de la falla cardíaca mediante la inhibición del intercambiador de sodio-calcio mitocondrial 91.
9 Conclusiones
Se ha determinado, que trastornos cardíacos como la hipertrofia cardíaca o la insuficiencia cardíaca, están asociados con alteraciones del sistema de fosforilación oxidativa, así como, con la reducción de los niveles de cardiolipina, junto con la desestabilización de los supercomplejos mitocondriales. Dichas disfunciones mitocondriales, también se han evidenciado, en distintos grados, en las dos subpoblaciones mitocondriales presentes en el músculo cardíaco. Las cardiopatías se caracterizan por exhibir una baja producción de ATP para los procesos de relajación y contracción del músculo cardíaco; es por ello, que disfunciones en la ATP sintasa, en el trasportador de ADP/ATP, y en el proceso de oxidación de ácidos grasos, han sido implicas en la aparición de insuficiencia cardíaca. Diferentes fármacos encargados de optimización la bioenergética mitocondrial, están siendo estudiados para ser utilizados en el tratamiento de diferentes afecciones cardíacas. Colombia cuenta con pocas investigaciones relacionadas con el tema, es por ello que conocer y entender los diferentes mecanismos involucrados en la generación de cardiomiopatías, permite seguir estimulando la creación de mecanismos, enfocados en el diagnóstico y prevención de patologías cardíacas, mediante la estabilización de la función mitocondrial. Todo lo anterior, es con el propósito de mejorar la calidad de vida de los pacientes y reducir los costos a largo plazo generados por dichas patologías.
Agradecimientos
Los autores agradecen a la Universidad EAFIT por la financiación de la Maestría y el préstamo de las instalaciones para la realización de los procesos investigativos. Así mismo, agradecemos al Doctor Luis Alejandro Gómez por su apoyo y por compartir todo su conocimiento con nosotros. Igualmente, gracias a cada una de aquellas personas que se involucraron en la revisión del presente trabajo

