










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista colombiana de Gastroenterología
versión impresa ISSN 0120-9957
Rev Col Gastroenterol vol.34 no.2 Bogotá abr./jun. 2019
https://doi.org/10.22516/25007440.239
Case report
Familial adenomatous polyposis and colorectal cancer prevention: A case report
1Médico Cirujano. Interno de Investigación del Centro de Investigación Radiológica, Universidad de Columbia. Nueva York, Estados Unidos
2Médico Cirujano. Interno de Posgrado del Hospital Dr. Miguel Pérez Carreño, Instituto Venezolano de los Seguros Sociales (IVSS). Caracas, Venezuela
Familial adenomatous polyposis (FAP) is a hereditary disease characterized by the growth of multiple colorectal epithelial adenomas. It is an autosomal dominant disorder caused by an APC gene defect. Degeneration to colorectal cancer is considered unavoidable in these patients if they do not receive adequate therapeutic management.
We present the case of a 25-year-old female patient consulted after a change in her evacuation pattern and abdominal pain. She had no relevant family history associated but based on results of paraclinical tests diagnosis of FAP was made for which therapeutic management was implemented. This is a case report with a literature review and update of the topic highlighting clinical issues related to recognition of the disease and issues that should be taken into consideration for the prevention of cancer in patients with FAP.
Keywords: Familial adenomatous polyposis; cannula
La poliposis adenomatosa familiar (PAF) es una enfermedad hereditaria caracterizada por el crecimiento de múltiples adenomas epiteliales de distribución colorrectal, de patrón autosómico dominante causado por el defecto del gen APC. La degeneración de cáncer colorrectal en estos pacientes se considera inevitable en caso de no recibir el manejo terapéutico adecuado.
Se presenta el caso de una paciente femenina de 25 años, quien acudió a consulta luego de presentar una modificación del patrón evacuatorio y dolor abdominal, sin antecedentes familiares asociados, por lo que se correlacionó con paraclínicos y se diagnosticó PAF, con la posterior implementación del manejo terapéutico. Se decidió hacer una revisión bibliográfica y actualización del tema resaltando los aspectos clínicos de reconocimiento de la enfermedad, así como las conductas a tomar en consideración para la prevención del cáncer en pacientes con PAF.
Palabras clave: Poliposis adenomatosa familiar; cáncer colorrectal; prevención
Introduction
Familial adenomatous polyposis (FAP) is inherited through transmission of an autosomal dominant pattern characterized by an APC gene defect located on the long arm of chromosome 5q21. 1-5 Only 25% to 30% of FAP patients do not have clinical or genetic evidence of the disease among family members. 6 The incidence of documented FAP in family records ranges from 1 in 7,000 to 1 in 16,000 live births and represents approximately 0.5% of all colorectal cancers. 4 The average age of onset is 16 years and the progression to colorectal cancer (CRC) occurs from the age of 40 to the age of 50 years with almost complete penetrance. 5,7
The appearance of hundreds or thousands of polyps on the rectal, colonic, duodenal and/or gastric mucosa is the primary manifestation of the disease. 8,9 Polyps have tubular, villous or mixed glandular histological structures, but the size of the polyps, rather than their structures, is the most significant predictive factor for the onset of cancer. 10 FAP usually has extracolonic manifestations such as gastric and small intestinal polyps. 1 Extraintestinal manifestations can include congenital hypertrophy of the retinal pigment epithelium, diffuse mesenteric fibrosis (desmoid tumors), osteomata of the lower jaw (in 90% of cases), skull and long bones (a phenotypic variant known as Gardner’s syndrome), and various dental abnormalities. 11 Neoplasms such as medulloblastoma (Turcot’s syndrome) can also occur in the central nervous system, thyroid glands, hepatobiliary system and adrenal glands. 7,8,12
The most important diagnostic approach to FAP is screening of patients with family histories even though it is believed that one third of all cases are due to de novo mutations. 13,4 Patients without any family history present great diagnostic challenges with negative prognostic implications. Surgical treatment aimed at preventing development of CRC is indicated when FAP is diagnosed, especially when the risk of onset is very high. 14 Development of CRC is inevitable when the disease follows its natural course in patients who do not undergo surgery. 1,14,15
Case presentation
The patient was a 25-year-old woman who had suffered from bloody stools and abdominal pain for a month prior to admission. Usually in the morning, she had one bloody stool without mucus or diarrhea followed by intense colic lasting for approximately one hour. Her defecation pattern changed from three/day to one/day.
Personal and Family Background
She was endoscopically diagnosed with chronic gastritis at the age of 13 when a single sessile polyp was found. One grandmother had died at age 69 from breast cancer. Her other grandmother was alive and had rheumatoid arthritis. Her mother was alive and had been diagnosed with uterine fibroids. The patient had no knowledge of any family history of CRC, polyps or FAP.
Physical Examination
The patient weighed 45 kg, was 168 cm tall, and had a body mass index (BMI)of 15.9. Her abdomen was flat, soft, depressible, painful on deep palpation of the lower hemiabdomen, with no signs of peritoneal irritation or visceromegaly and with sounds of liquid and air. Rectal examination was not painful and found normal muscle tone and temperature of the anal sphincter and rectal walls with multiple palpable masses of different diameters. The rectal ampulla contained pasty stool and the tip of the glove showed a small amount of blood. The rest of the evaluation was within normal limits.
Paraclinical Tests
A complete blood count and blood chemistry were normal. In addition, barium enema abdominal radiography found alterations of the colonic frame with accumulation of secretion (Figure 1). In the rectosigmoidoscopy, more than 100 polyps of sizes ranging from 3 mm to 5 cm were observed from the distal ascending colon to the rectum. Some were pediculate and others were sessile, tubular and villous in appearance (Figures 2 and 3).
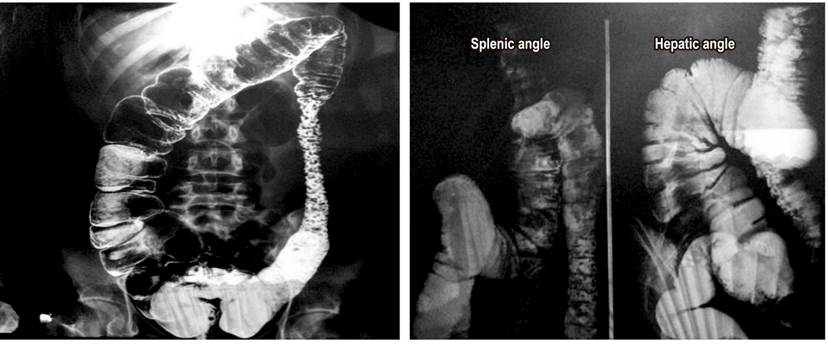
Figure 1 Abdominal barium enema radiography in standing anteroposterior projection shows evidence of colonic wall thickening, irregular dilations of the colonic frame, increased amounts of heterogeneous secretions in the ascending colon, hepatic angle, descending colon and sigmoid colon with multiple polypoid images and correlative signs with collar ulcers.
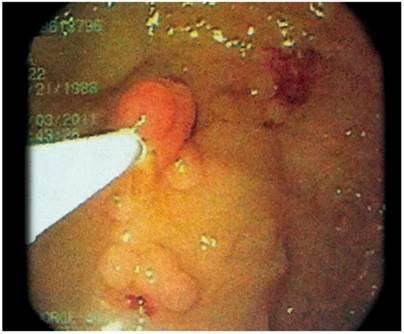
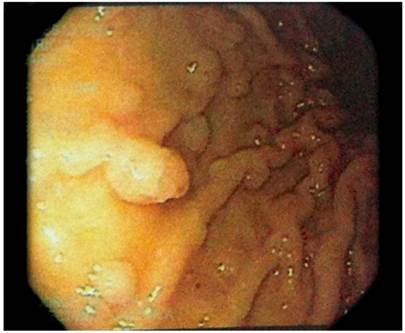
A gastroduodenoscopy showed white cottony points in the second portion of the duodenum with multiple sessile polypoid lesions in the corpus and fundus (Figure 4).
The histopathological study shown in Figure 4 reported that:
The mucosa of the sigmoid colon had tubular adenoma with high grade focal dysplasia (10%).
There were tubular villous polyps in the transverse colon polyps and adenoma with a predominance of the adenomatous component in 80% of the material examined plus areas of high grade focal dysplasia.
Hyperplastic oxyntic gland adenoma with dilation of glandular crypts and chronic mucosal inflammation in the gastric corpus.
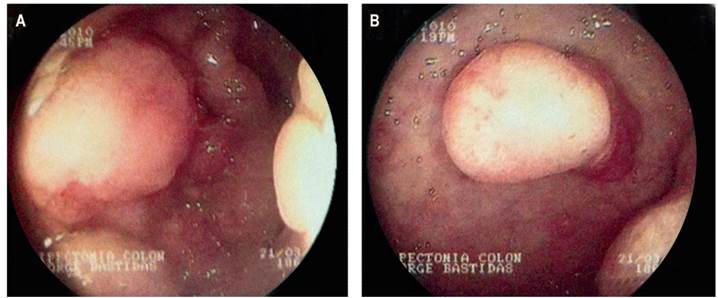
Figure 4 Gastroduodenoscopy. (A) Fundus (B) Corpus with multiple sessile polyps of various diameters.
According to the clinical and the complementary evidence, a diagnosis of FAP was established.
Treatment and Evolution
A total colectomy plus resection of the upper two thirds of the rectum with an ileorectal anastomosis was indicated. First intervention, a protective ileostomy was placed. Pathological analysis of ileal, colonic and rectal tissue obtained showed countless polypoid structures mostly consisting of pediculate and sessile tubulovillous adenomas with evidence of high-grade dysplasia seated on the mucosa with multifocal hyperplastic changes. This intervention evolved favorably.
The patient was re-admitted six months later for scheduled end to end anastomosis. Paralytic ileus developed in the immediate postoperative period but was successfully treated medically. In the year after surgery, the patient had three episodes of intestinal obstruction that merited surgical resolution. In the last episode, an intra-abdominal abscess in the right iliac fossa led to placement of a Bogotá bag which required hospitalization for 8 months. The patient’s condition evolved favorably, and she is currently is in good general condition with regular intestinal transit and without any other type of compromise.
Discussion
Although FAP has been disease studied and described, prevention and early diagnosis are key for the prognosis, and in this lies the relevance of this review considering that all untreated cases evolve toward in malignancy. 13 Diagnosis is based on clinical and endoscopic evidence. 12 The most common symptom is blood in the stool (occult or visible) mainly because polyps bleed into their stroma as more and more polyps progressively appear. 1,8 Other possible symptoms are flatulence, changes in defecation pattern, and post-defecation colic in the lower hemiabdomen. 7,8 Anorectal examination may show polyps if they prolapse through the rectum. 1 Family history is also of great importance, so the patient should be asked about family history of colonic polyps and/or colon cancer. 15 Nevertheless, about one third of these patients do not have any related family history, as in the case of our patient.
The diagnosis is established by clinical criteria by means of a barium enema and a total colonoscopy which can show evidence of larger lesions and be used to take biopsy samples to rule out malignancy. 16 This differentiates the disease into either its classical variation of more than 100 polyps or into its attenuated variation with less than 100 polyps. 7 A search for extracolonic findings, especially duodenal polyps and epithelial hyperpigmentation of the retina, is necessary. The latter is observed in 83% of families with FAP. 7,17 Screening of relatives and identification of the APC gene are both important although genetic study of this patient was not possible for socioeconomic reasons. 2,14
Surgery is the fundamental pillar for management of FAP, particularly when there is a mutation between codons 1251 and 1309 which express the most serious phenotype. 13 Individualization of each patient and genotype-phenotype correlation are important for choosing among surgical options which include total proctocolectomy with permanent ileostomy, colectomy with ileorectal anastomosis (IRA), and proctocolectomy with ileal pouch-anal anastomosis (IPAA). The first approach is not performed prophylactically and is indicated in patients with extensive rectal polyposis, cancer of the distal rectum and for those for whom follow up is impossible. On the other hand, the IRA colectomy and the IPAA proctocolectomy are prophylactic techniques carried out in asymptomatic patients who have been identified as being at risk by genetic tests or predictive colonoscopy. However, the risk of developing cancer in the preserved rectal portion after 20 years is 25%, so periodic monitoring must be particularly strict. 13,18
Medical treatment can be considered for easily monitored patients with the attenuated variant with less than 20 rectal polyps whose size is less than 5 mm. 19,20 The use of non-steroidal anti-inflammatory drugs (NSAIDs) such as sulindac and celecoxib has been shown to be the most efficient at reducing the amount and size of colorectal adenomas characterized by high levels of cyclooxygenase 2 (COX-2) expression. 20,21 Their use is accepted as adjuvant therapy to surgical treatment but not as an alternative. 22 Surgical resolution is controversial in asymptomatic patients for whom intervention is recommended at the end of adolescence. 14
In at-risk individuals with evidence of polyposis without PAF or its attenuated variant, annual colonoscopy or flexible sigmoidoscopy is suggested from onset of puberty. 5,10,19 Colonoscopic follow up of patients who have undergone IRA colectomy should be initiated six months after surgery and thereafter be done once a year. The initial follow-up is the same for proctocolectomy patients with IPAA with subsequent follow-up every two or three years. 18,22. This periodicity is conditioned by baseline and follow up findings of the number, size and histology of adenomas as well findings of any symptoms or mutations in the APC gene. 10
The close relationship of exhaustive study of each patient with correct therapy choices and good prognoses demonstrates the crucial role of permanent analysis of diagnostic guidelines and constant investigation of the subject since about 30% of patients with this disease have no relevant family history. 4 As in the case presented, absence of family history can result in diagnosis at a later age after symptoms develop which implies increased risks of morbidity, mortality and development of CRC. 4 Additional studies of the genetics and epidemiology of patients without family history could improve the bases of diagnosis and clinical management.
Referencias
1. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. https://doi.org/10.1186/1750-1172-4-22 [ Links ]
2. Bisgaard ML, Ripa R, Knudsen AL, Bülow S. Familial adenomatous polyposis patients without an identified APC germline mutation have a severe phenotype. Gut. 2004;53(2):266-70. https://doi.org/10.1136/gut.2003.019042 [ Links ]
3. Cao X, Hong Y, Eu KW, Loi C, Cheah PY. Singapore familial adenomatous polyposis (FAP) patients with classical adenomatous polyposis but undetectable APC mutations have accelerated cancer progression. Am J Gastroenterol. 2006;101(12):2810-7. https://doi.org/10.1111/j.1572-0241.2006.00842.x [ Links ]
4. Truta B, Allen BA, Conrad PG, Weinberg V, Miller GA, Pomponio R, et al. A comparison of the phenotype and genotype in adenomatous polyposis patients with and without a family history. Fam Cancer. 2005;4(2):127-33. https://doi.org/10.1007/s10689-004-5814-0 [ Links ]
5. Kobayashi H, Ishida H, Ueno H, Hinoi T, Inoue Y, Ishida F, et al. Association between the age and the development of colorectal cancer in patients with familial adenomatous polyposis: a multi-institutional study. Surg Today. 2017;47(4):470-475. https://doi.org/10.1007/s00595-016-1398-1 [ Links ]
6. Dalavi SB, Vedpalsingh TH, Bankar SS, Ahmed MHS, Bhosale DN. Familial adenomatous polyposis (FAP): a case study and review of literature. J Clin Diagnostic Res. 2015;9(3):PD05-PD06. https://doi.org/10.7860/JCDR/2015/11636.5696 [ Links ]
7. Kennedy RD, Potter DD, Moir CR, El-Youssef M. The natural history of familial adenomatous polyposis syndrome: A 24-year review of a single center experience in screening, diagnosis, and outcomes. J Pediatr Surg. 2014;49(1):82-86. https://doi.org/10.1016/j.jpedsurg.2013.09.033 [ Links ]
8. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101(2):385-98. https://doi.org/10.1111/j.1572-0241.2006.00375.x [ Links ]
9. Arnason T, Liang WY, Alfaro E, Kelly P, Chung DC, Odze RD, et al. Morphology and natural history of familial adenomatous polyposis-associated dysplastic fundic gland polyps. Histopathology. 2014;65(3):353-362. https://doi.org/10.1111/his.12393 [ Links ]
10. Winawer SJ, Zauber AG, Fletcher RH, Stillman JS, O’Brien MJ, Levin B, et al. Guidelines for Colonoscopy Surveillance After Polypectomy: A Consensus Update by the US Multi-Society Task Force on Colorectal Cancer and the American Cancer Society. Gastroenterology. 2006;130(6):1872-85. https://doi.org/10.1053/j.gastro.2006.03.012 [ Links ]
11. Smith WG, Kern BB. The nature of the mutation in familial multiple polyposis: papillary carcinoma of the thyroid, brain tumors, and familial multiple polyposis. Dis Colon Rectum. 1973;16(4):264-71. [ Links ]
12. Durno C, Monga N, Bapat B, Berk T, Cohen Z, Gallinger S. Does Early Colectomy Increase Desmoid Risk in Familial Adenomatous Polyposis? Clin Gastroenterol Hepatol. 2007;5(10):1190-4. https://doi.org/10.1016/j.cgh.2007.06.010 [ Links ]
13. Tudyka VN, Clark SK. Surgical treatment in familial adenomatous polyposis. Ann Gastroenterol Q Publ Hell Soc Gastroenterol. 2012;25(3):201-6. [ Links ]
14. Win AK, Walters RJ, Buchanan DD, Jenkins MA, Sweet K, Frankel WL, et al. Cancer risks for relatives of patients with serrated polyposis. Am J Gastroenterol. 2012;107(5):770-8. https://doi.org/10.1038/ajg.2012.52 [ Links ]
15. Balaguer Prunés F, Castells i Garangou A. Clínica de alto riesgo de cáncer colorrectal: un nuevo concepto de prevención. Gastroenterol Hepatol Contin. 2007;6(6):289-94. [ Links ]
16. Iwama T, Tamura K, Morita T, Hirai T, Hasegawa H, Koizumi K, et al. A clinical overview of familial adenomatous polyposis derived from the database of the Polyposis Registry of Japan. Int J Clin Oncol. 2004;9(4):308-316. https://doi.org/10.1007/s10147-004-0414-4 [ Links ]
17. Cordero-Fernández C, Garzón-Benavides M, Pizarro-Moreno A, García-Lozano R, Márquez-Galán JL, López Ruiz T, et al. Gastroduodenal involvement in patients with familial adenomatous polyposis. Prospective study of the nature and evolution of polyps: evaluation of the treatment and surveillance methods applied. Eur J Gastroenterol Hepatol. 2009;21(10):1161-7. https://doi.org/10.1097/MEG.0b013e3283297cf2 [ Links ]
18. Brandão C, Lage J. Management of Patients with Hereditary Colorectal Cancer Syndromes. GE Port J Gastroenterol. 2015;22(5):204-12. https://doi.org/10.1016/j.jpge.2015.06.003 [ Links ]
19. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW; et al. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am J Gastroenterol. 2015;110(2): 223-63. https://doi.org/10.1038/ajg.2014.435 [ Links ]
20. Kim B, Giardiello FM. Chemoprevention in familial adenomatous polyposis. Best Pract Res Clin Gastroenterol. 2011;25(4-5):607-22. https://doi.org/10.1016/j.bpg.2011.08.002. [ Links ]
21. Bresalier RS. Primary chemoprevention of familial adenomatous polyposis with sulindac: More questions than answers. Gastroenterology. 2002;123(1):379-81. https://doi.org/10.1053/gast.2002.1230379 [ Links ]
22. Navarro M, González S, Iglesias S, Capellá G, Rodríguez-Moranta F, Blanco I. Síndrome de poliposis hiperplásica: diversidad fenotípica y asociación a cáncer colorrectal. Med Clin (Barc). 2013;141(2):62-6. https://doi.org/10.1016/j.medcli.2012.04.024 [ Links ]
Received: March 23, 2018; Accepted: June 13, 2018
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
