











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
Castleman disease (CD) is a non-clonal lymphopro liferative disorder. More than a disease, it constitutes a heterogenous group of very rare entities with a broad range of clinical manifestations 1. The first case was reported in 1956 as part of a series of patients with few or no symptoms, but with a solitary mediastinal ganglion 2. There are three major recognized variants of CD: hyaline vascular (HV), plasma cell and human herpesvirus-8 (HHV8) positive. The initial report by Benjamin Castleman was of the HV subtype, in which the architecture of the lymphatic glands is characterized by lymphoid follicles with atrophic or "regres sive," often hyalinized, germinal centers, made up mainly of residual follicular dendritic cells and prominent mantle zones containing small lymphocytes. The follicular dendritic cells are arranged concentrically and have an "onion skin" appearance. The plasma cell variant is the most common subtype of the multicentric disease (75%) 3. The coexis tence of hyalovascular and plasma cells is characteristic of a multicentric CD subtype associated with HHV8 4.
In 1990, Anhalt et al. reported a clinical entity called "paraneoplastic pemphigus" (PNP). Subsequently, Nguyen et al. described it as a heterogenous autoimmune syndrome affecting various internal organs, since its pathophysiology is not limited to autoantibodies directed against adhesion molecules. Paraneoplastic pemphigus is closely related to benign or malignant tumors. The most frequently reported malignant tumors are lymph tissue and hematological tumors (B cell lymphoma, chronic lymphocytic leukemia, Castleman disease, Waldenstrom macroglobulinemia and thymoma) 5. We present the case of a male patient with oral and genital ulcers in whom PNP was diagnosed as an atypical presentation of multicentric CD.
Case presentation
A 52-year-old male patient consulted due to a six-month history of asthenia, adynamia, hyporexia, a weight loss of approximately 18 kg, painful mouth and genital ulcers, and red, itchy eyes. He received empirical antibiotic treatment with penicillin at another institution and was then treated with prednisone and thalidomide for suspected Behçet's disease, without remission of the clinical manifestations. The physical exam showed conjunctival hyperemia, oral ulcers not affecting the gingiva (Figure 1A) and genital ul cers involving the scrotum and foreskin (Figure 1B), along with a nonreducible left varicocele. Complementary studies showed: a complete blood count with thrombocytosis at 506 x 109/L, leukocytes at 6.4 x 109 g/L and hemoglobin at 150 g/L. Blood chemistries reported hyperuricemia (562 / μmol/L), an accelerated erythrocyte sedimentation rate (80 mL/hour), elevated C-reactive protein at 80 milligrams (mg)/L, and negative antinuclear antibodies, antineutrophil cytoplasmic antibodies, anti-HHV-8 antibodies, and anti-human immunodeficiency 1 and 2 antibodies. He was seen by rheumatology who ordered a pathergy test which was reported as negative, and an ophthalmology exam which showed no signs of anterior or posterior uveitis, ruling out the possibility of Behçet' s disease. A bone marrow biopsy showed no tumor infiltration. Imaging studies included an abdominal ultrasound which showed a retroperitoneal mass, leading to a computerized axial tomography of the abdomen with contrast where a solid 94 x 51 millimeter mass with heterogenous density and multiple microcalcifications was found in the retroperitoneum, to the right of the midline, with ventral displacement and compression of the left renal vein (which explained the nonreducible ipsilateral varicocele found on physical exam), along with multiple adjacent enlarged retroperitoneal lymph nodes (Figure 2). The tomographic study was extended to the chest, finding multiple posterior mediastinal paraesophageal adenopathies extending into the retrocrural space, the largest measuring 23 x 34 mm. A tomography-guided biopsy of the retroperitoneal mass was taken, which was reported as the HV variant of Castleman disease (Figure 3A), with the following immunohistochemical specifications: CD3, CD20, KI67, and CD21 positive, and BCL6 and CD30 negative. A biopsy was also taken of the fore skin lesion (Figure 3B), with a histopathological report describing the almost total loss of the epidermis with intraepidermal separation secondary to acantholysis, the presence of numerous isolated and clustered acantholytic cells with hyperchromatic nuclei and homogenous cyto plasm, necrotic keratinocytes and basal vacuolization. In addition, immunological fluorescence was found in the epidermal intercellular spaces, with monoclonal antibodies directed against immunoglobulin G and complement factor 3. Considering the histopathology report, positive direct immunofluorescence and macroscopic skin findings previ ously described, in the presence of CD, the existence of PNP was solidly suspected, and therefore anti-desmoglein 1 and 3, anti-desmoplakin 1 and antienvoplakin antibod ies, along with bullous pemphigoid antigen, were ordered, which were positive. Indirect immunofluorescence on monkey esophagus or rat bladder transitional epithelium was not ordered because it was not available, but based on the clinical, imaging, immunological and anatomopathological findings previously described, PNP-associated HV variant multicentric Castleman disease was diagnosed. He received chemotherapy with cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) without achieving partial or total remission of the oral and genital ulcers diagnosed as PNP.
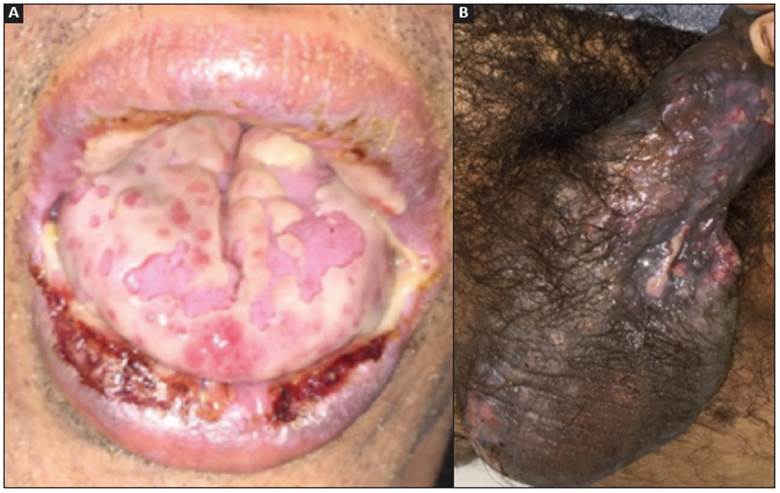
Figure 1 A. Severe stomatitis. Erosive cheilitis with serosanguinous scabs. The tongue has erosions covered by whitish membranes. B. Ulcerated lesions covered with a whitish membrane on the scrotum and foreskin
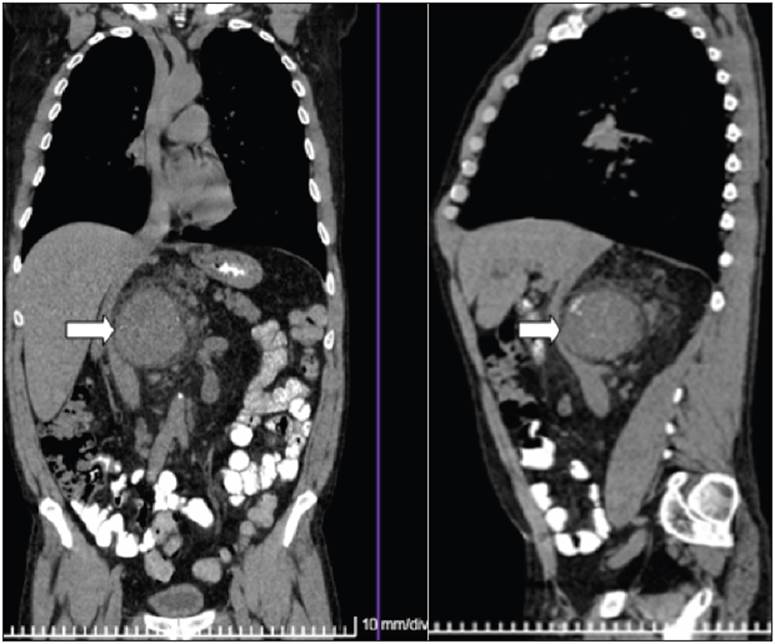
Figure 2 Abdominal CAT . A solid retroperitoneal mass with heterogenous density and multiple microcalcifications to the right of the midline due to Castleman disease in a patient with paraneoplastic pemphigus
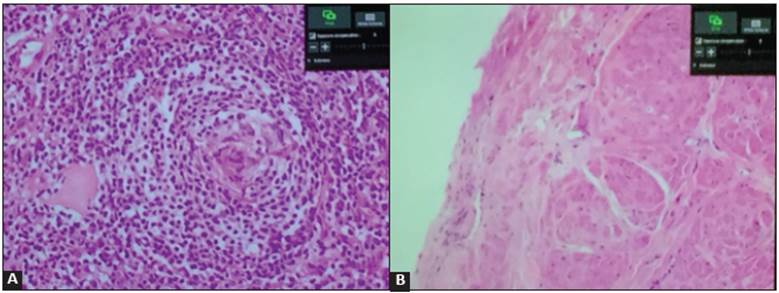
Figure 3 A. Histopathology of a retroperitoneal mass showing small lymphoid follicles with concentric arrangement of the lymphocytes, “onion skin” appearance of the angiofollicular hyperpla sia, compatible with hyaline vascular variant Castleman disease, (hematoxylin and eosin 40x). B. Histopathology of a skin lesion showing almost total loss of the epidermis with intraepidermal separation secondary to acantholysis and accompanying necrosis compatible with pemphigus (hematoxylin and eosin 40x).
Discussion
Castleman disease is a rare lymphoproliferative disorder which was first described more than 60 years ago as a be nign, localized mass 6. From a clinical perspective, this disease has been divided into two subtypes: unicentric CD (UCD) and multicentric CD (MCD). There is a difference in the clinical manifestations of the stated subtypes, as UCD generally tends to have few symptoms and its most frequent histological variant is HV, accounting for 80-90% of the cases, while MCD includes a plasma cell and a mixed type, which often cause systemic signs and symptoms like fever, weight loss, hypoproteinemia, systemic lymphadenopathy, decreased kidney function, and even POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, mono clonal protein and skin changes) 6-7. It should be noted that the HV histological variant has a unicentric clinical presentation in approximately 90% of cases. However, it may be multicentric, as in our patient, who had retroperitoneal lymphadenopathies adjacent to the tumor mass and in the mediastinum. The multicentric form is aggressive and may be associated with conditions like POEMS syndrome, amyloidosis, Kaposi's sarcoma, kidney failure and HHV-8 and HIV infection. The most common locations of CD in the body are thoracic (60%), cervical (14%), abdominal (11%) and axillary (4%) 8. In our specific case, we found the prominent tumor site in the retroperitoneum, which is not one of the most common sites, accounting for approximately 3% of cases. However, the histological subtype was HV, which is the most frequent in most of the series reviewed, such as that of Kawamura et al. 9 who reported that the HV subtype was found in 87% of 132 cases of retroperitoneal CD in Japanese people.
In light of the fact that the cardinal clinical elements of our case were in the skin, with a diagnosis of PNP, it should be noted that approximately 18% of these cases are associated with CD. However, there are few reported cases of PNP associated with CD 10. Paraneoplastic pemphigus is an autoimmune mucocutaneous disease characterized by severe stomatitis, polymorphic skin rashes and associated underlying neoplasms. Its histopathological characteristics are dyskeratotic epithelial cells with acantholysis and typical immunofluorescence. Paraneoplastic pemphigus has been known for having a poor prognosis with mortality rang ing from 75 to 90%, and a mean survival of less than one year 11. A combination of criteria are recommended for diagnosing PNP, including painful mucosal erosions and polymorphic skin rashes, histopathological characteristics of intraepidermal acantholysis, dyskeratosis and vacuole interphase dermatitis; direct immunofluorescence findings of intercellular epidermal IgG and complement; serum antibodies detected by indirect immunofluorescence that bind to superficial stratified squamous epithelial cells, as well as to simple, columnar and transitional epithelium; and, finally, serum immunoprecipitation with a four-protein complex (desmoplakin I -250 kD-, bullous pemphigoid antigen-230 kD-, desmoplakin II and envoplakin -210 kD-, and periplakin -190 kD). These diagnostic criteria were subsequently reviewed by Camisa and Helm who divided them into major and minor signs: the first include polymorphic mucocutaneous rashes, concurrent neoplasms and serum antibodies with a specific immunoprecipitation pattern; while the minor signs include histological evidence of acantholysis, direct immunofluorescence showing inter cellular and basal membrane staining, and an indirect immunofluorescence test using rat bladder epithelium. Three major, or two major and two minor, criteria are needed to diagnose PNP. In the specific case of our patient, we had three major and two minor criteria to support our diagnosis. The most effective treatment for PNP is resection or resolu tion of the underlying neoplasm, as has been shown when it is associated with CD. Mortality due to bronchiolitis obliterans, which is the most severe complication, is quite low, possibly due to early removal of the Castleman tumor and administration of immunoglobulin 11,12. High-dose systemic corticosteroids such as prednisone (1.0-1.5 mg/ kg/day) and immunosuppression with azathioprine and mycophenolate have also been recommended for treatment; rituximab has been proposed as well, since it acts by reduc ing the B cells responsible for producing the antibodies that cause the skin lesions 13. Although our patient received high-dose steroids and a CHOP therapy cycle, he showed no improvement in the mucocutaneous lesions.
Castleman disease treatment is aimed at the pathogenesis of the suggested disease. Surgical excision is indicated for unicentric disease (either the HV or plasma cell variant), but is rarely used for the multicentric form, which is our specific case, while cytoreduction therapy may be used. The most commonly used chemotherapy is the CHOP regimen which was administered to our patient, or cyclophosphamide, vincristine, doxorubicin and dexamethasone (CVAD) 14. Some studies have shown benefits with radiation therapy and immunomodulators like steroids, alpha interferon, trans retinoic acid and thalidomide 14-16. Other treatments are monoclonal antibodies which include those aimed at inhibiting interleukin 6, such as siltuximab and altizumab, as well as the anti-CD20, rituximab 17. Although anti-CD20 monoclonal antibodies were indicated in our patient, since they were one of the immunohistochemical markers expressed, we were not able to use this therapeutic strategy as rituximab was temporarily unavailable in our setting. Therefore, interferon was added to the treatment to reduce the burden of CHOP therapy. Various antiviral medications like acyclovir, ganciclovir, cidofovir and valganciclovir have also been used to treat the viremia associated with CD 14,16,18.
During his stay in our institution, our patient received follow up and comprehensive treatment from a multidisciplinary group. However, he developed cellulitis on his right arm and, due to the degree of immunodepression, despite broad spectrum antibiotic treatment, he progressed ominously to sepsis and then to septic shock and died during his stay in the polyvalent intensive care unit.
Conclusion
Mucocutaneous lesions are a frequent finding in medical practice, with an etiological spectrum spanning infectious processes, immunological diseases and neoplasms. When they are resistant to antimicrobial and immunosuppressive treatment, we should consider that they might be part of the clinical spectrum of a paraneoplastic syndrome. We presented the case of a patient with PNP associated with HV variant CD, which usually has a benign course in the unicentric clinical form. However, our patient had the multicentric type, with an ominous clinical progression, and histopathology was essential for his diagnosis.

