











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
Protoanemonin, (1), 5-methylene-2(5H)-furanone, is a secondary metabolite produced by numerous species of Ranunculaceae (Figure 1). It is formed by enzymatic hydrolysis of its precursor, the inactive glycoside ranunculin, as a consequence of tissue impairment [1]. Pseudomonas sp. B13 and Pseudomonas reinekei MT1 can produce protoanemonin as a product in the biodegradation of organochloride xenobiotics [2-4].
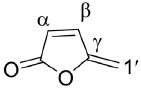
Protoanemonin has shown diverse biological activities. This furanone is present in several plants used in Chinese folk medicine for the treatment of inflammatory processes and for the prevention and treatment of various types and stages of cancer [5,6]. It possesses antibacterial [7,8], antifungal [9], antimutagenic [10], and potential antitumor properties [11,12].
Undoubtedly, this range of biological activities is due to its chemical reactivity and high lipophilicity. Protoanemonin has an a,β-unsaturated lactone moiety, which can interact with several nucleophilic groups such as thiols or amines in a Michael-type reaction, that could induce cytotoxic effects [13]. According to the above, the protoanemonin exhibits a moderate cytotoxicity against A549, NIH 3T and SK-OV-3 cancer cell lines. Also, it showed weak cytotoxicity against MCF-7 human breast cancer line. Nevertheless, the 5-bromo-5-methyl-2(5H)-furanone derivative was reported to exert the strongest effect against the MCF-7 cell line; besides, it is useful in the suppression of HepG2 and A549 human neoplastic cell lines. This Br-derivative also induces apoptosis and cell cycle arrest [14]. According to these results, it seems that the inclusion of a Br atom in the protoanemonin nucleus enhances its cytotoxic activity. Considering this, we decided to synthesize the known bromo furanones: (Z)-4-Bromo-5-(bromomethylene)-furan-2(5H)-one (3), 5-(Dibromomethylene)-2(5H)-furanone (4), and (E)-5-(Bromomethylene)-2(5H)-furanone (5), Figure 2, and compare their cytotoxic activities with that of protoanemonin (1), which was synthesized by a new reaction.
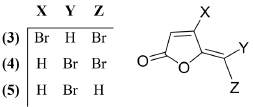
Figure 2 Brominated furanones: (Z)-4-Bromo-5-(bromomethylene)-furan-2(5H)-one (3), 5-(Dibromomethylene)-2(5H)-furanone (4), and (E)-5-(Bromomethylene)-2(5H)-furanone (5).
Several approaches to the synthesis of (1) have been published. Among them, protoanemonin was formed by the reaction between acetic anhydride and acetylacrylic acid [15]. In another approach, the a-angelica lactone, obtained from levulinic acid, was used as synthetic precursor [16]. It was also produced by the photo-oxygenation of 5-hydroxymethylfurfural [17]. Furthermore, the synthesis of the protoanemonin from 2-deoxy-D-ribose also has been reported [18]. One-pot synthesis of protoanemonin was achieved by a catalyst system composed by the combination of 10% Pd/C with CuI, PPh3, and Et3N coupling of (Z)-3-iodoacrylic acid and trimethylsilyl acetylene [19].
The aim of the present study was to synthesize protoanemonin and brominated furanones (3) to (5) to investigate their potential cytotoxicity against PC-3 and U-251 cancer cell lines. We describe the synthesis of (1) from levulinic acid, which was achieved by the combination of two methods previously published. Initially, the mixture of brominated furanones (3)-(5) was obtained from levulinic acid [20]. Then, (1) was obtained from (3) by reduction with zinc [21]. Cytotoxic activity of protoanemonin and brominated furanones (3)-(5) was evaluated against PC-3 and U-251 human cancer cell lines by using colorimetric SRB bioassay (Figure 3).
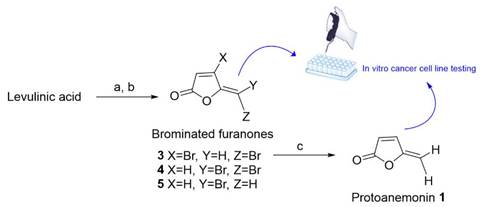
Materials and methods
General experimental procedure
Uncorrected melting points were taken on a Fisher Johns apparatus. The 1D and 2D nuclear magnetic resonance (NMR) spectra were recorded by Varian VXR-300 and Bruker Avance 111-400 spectrometers with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are expressed in ppm, concerning the solvent signals. Mass spectra were recorded on a JEOL AX505 mass spectrometer. Silica gel 70-230 (Merck Mexico) was used for column chromatography. Preparative TLC (PTLC) was performed on 2 mm pre-coated plates of SILG/UV254 (Merck Mexico) and thin-layer chromatography was carried out on aluminum SILG/UV254 sheets (Merck Mexico).
Chemicals
The substances used in this work were levulinic acid > 98%, bromine ≥ 99%, zinc powder > 98%, hydroquinone ≥ 99%, tetrahydrofuran, isopropyl ether, glacial acetic acid, sulfuric acid, ethylic ether, sodium sulfate, and celite (all were purchased from Sigma Aldrich, USA). Hexane, dichloromethane, ethyl acetate and methanol, used in chromatography, were industrial grade double distilled.
Synthesis of furanones (3)-(5)
A solution of levulinic acid (10 g, 0.086 mol) in methanol (80 mL, dried by storage over 3 Å molecular sieves, 20% w/v, after distillation) was stirred at 20 °C in bath of water, while bromine (26 g, 0.16 mol) was slowly added at a rate which maintained the temperature of the solution at 30 °C. After 2.5 h, the solution was refluxed for 1h and methanol was removed under reduced pressure. The residue was stirred with ether (80 mL) and water (25 mL) while an excess of sodium bicarbonate solution was added. The ether layer was dried, and ether was distilled off [22].
The concentrated sulfuric acid (20 mL) was added to the crude mixture (2) (6.8 g) at room temperature, and the reaction was heated in an oil bath at 90-100 °C for 15 min. The mixture was cooled to room temperature and slowly poured on to crushed ice. The resulting solution was extracted with dichloromethane (3 x 60 mL). The extracts were washed with water, dried over Na2SO4, and evaporated to obtain an oily product. Compounds (3), (4) and (5) were purified as isolated products from the resulting oil by column chromatography (CC), using hexane-ethyl acetate 99:1. Each compound was washed with isopropyl ether. Structure identities of compounds (3)-(5) were established by comparison of their physical and spectroscopic data previously reported in the literature [20].
Synthesis of protoanemonin (1)
First, furanone (3) (100 mg, 0.39 mmol) was dissolved in anhydrous THF (5 mL). Then, activated zinc powder (1 eq) and few crystals of hydroquinone were added. After 20 min of mixture sonication, two drops of glacial acetic acid were added. The mixture was maintained in ultrasound irradiation, and CCF monitored the proceeding reaction at 15 min intervals. After 60 min, the reaction was completed, and the product was filtered over celite with methanol. Protoanemonin (1) was purified by preparative TLC eluting with hexane-ethyl acetate 70:30.
Cell lines
The glioblastoma (U-251) and prostate adenocarcinoma (PC-3) human tumor cell lines were supplied by the National Cancer Institute (NCI, Maryland, USA).
Antiproliferative assay
The antiproliferative activities of protoanemonin and brominated derivatives were evaluated against prostate adenocarcinoma (PC-3) and glioblastoma (U-251) cell lines, which were determined by using the protein-binding dye sulforhodamine B (SRB) in a microculture method [23]. This assay is efficient and highly sensitive to measure drug-induced cytotoxicity. Its staining is not cell-line dependent, but it is independent of cell metabolic activity, and the end-point measurement is not time-critical. Besides, it has been adopted for routine use in the National Cancer Institute for in vitro antitumor evaluation. The cells were cultured in RPMI 1640 medium, supplemented with 10% (v/v) inactivated fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin sulfate, 2 µg/mL glutamine, and 0.25 µg/mL amphotericin B. The cells were maintained in a humidified atmosphere (95% humidity) with 5% CO2 at 37 °C for 24 h prior to the addition of experimental compounds. The viability of the cells used in the experiments exceeded 95%, as determined by a trypan blue assay. The cells were removed from the tissue culture flasks by treatment with trypsin and diluted with fresh media. Cell suspensions (100 µL), containing 5000 or 7500 cells per well, were pipetted into 96 well plates (Corning, NY) and incubated at 37 °C for 24 h in a 5% CO2 atmosphere to reach exponential growth. Then, a 100 µL aliquot of the test compounds or positive control was added to each well. All tested substances were dissolved in DMSO at a maximum concentration of 0.5%, at which DMSO was totally innocuous. The cultures were exposed to the compounds for 48 h. After the incubation period, the cells were fixed in situ by adding 50 mL of cold 50% aqueous trichloroacetic acid (TCA) and incubated at 4 °C for 60 min. The supernatant was discarded, and the plates were washed with tap H2O and air-dried. The addition of 0.4% SRB stained the TCA-fixed cells. Then, free SRB solution was removed by washing with 1% v/v aqueous acetic acid. The plates were then air-dried, and the bound dye was solubilized by the addition of 10 mM unbuffered Tris base (100 µL). The plates were placed on a shaker for 5 min, and the absorption was determined at 515 nm by using an Ultra microplate reader (ELx808, Bio-Tek Instruments, Inc.Vermont, USA). Experiments were performed in triplicate. The concentration-response curve was plotted for each compound. Results were expressed as the concentration giving 50% inhibition (IC50) and were estimated via linear regression.
Results and discussion
The bromination of levulinic acid in methanol afforded an oleum 58:35:7 mixture of brominated products 2a / 2b / 2c in 71% yield (Figure 4). These products could be readily isolated by CC, but the crude mixture, (2), was used without purification for synthetic purposes. Identity of brominated products was determined by 1H NMR spectroscopy; the chemical shifts and coupling constants of compounds 2a-2c corresponded with published data [20,22]: Methyl 3,5-dibromo-4-oxopentanoate (2a) 1H NMR (CDCl3, 300 MHz) δ: 2.97 (1H, dd, J=16.9, 6.4 Hz, -CHH-), 3.30 (1H, dd, J=16.8, 8 Hz, -CHH-), 3.65 (3H, s, OCH3), 4.34 (1H, d, J=12.9 -CHHBr), 4.39 (1H, d, J=12.8, -CHHBr), 5.01 (1H, t, J=6.3, -CHBr-); methyl 5-dibromo-4-oxopentanoate (2b) (CDCl3, 300 MHz) δ: 2.90 (2H, t, J=6.5, -CH2-), 3.08 (2H, t, J=6.5, -CH2-), 3.67 (3H, s, -OCH3), 5.89 (1H, s,-CHBr2-), and methyl δ-bromo-4-oxopentanoate (2c) (CDCl3, 300 MHz) δ: 2.71 (2H, t, J=6.6, -CH2-), 2.98 (2H, t, J=6.7, -CH2-), 3.64 (3H, s, -OCH3), 3.95 (1H, s,-CH2Br-).
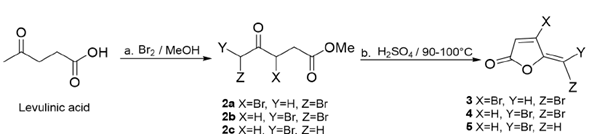
Thus, crude 2a/ 2b/ 2c mixture treated with sulfuric acid at 90-100 °C afforded bromo furanones (3), (4) and (5) in 41%, 12%, and 3% yield, respectively.
(Z)-4-bromo-5-(bromomethylene)-furan-2(5fl)-one (3) [20]. White crystals: mp 82 - 83 °C; 1H NMR (CDCl3, 300 MHz) δ: 6.41 (1H, s, 5-CHBr), 6.50 (1H, s, H-3). HPLC purity of 99.9%.
5-(dibromomethylene)-2(5fl)-furanone (4) [20]. Orange crystals: mp 124 - 126 °C, 1H NMR (CDCl3, 300 MHz) δ: 6.42 (1H, d, J=5.6 Hz, H-3), 7.68 (1H, d, J=5.6 Hz, H-4). HPLC purity of 99.2%.
(E)-5-(bromomethylene)-2(5fl)-furanone (5) [20]. White crystals: mp 75 - 76 °C, 1H NMR (CDCl3, 300 MHz) δ: 6.12 (1H, s, 5-CHBr), 6.33 (1H, d, J=5.4 Hz, H-3), 7.40 (1H, d, J=5.4 Hz, H-4). HPLC purity of 100 %.
Our target protoanemonin compound, (1), was settled by replacing the bromine atoms in furanone (3) by hydrogen, through smooth sonication with zinc dust and two drops of glacial acetic acid in THF during a short time (60 min) and at a yield of 35%. This procedure is based on the Sorg method to obtain halogen-free γ-alkylidenebutenolides [21]. Synthesis of protoanemonin was achieved using a three-step procedure: first, a bromo keto methyl ester mixture was obtained from a brominating reaction of levulinic acid; second, the acid cyclization of methyl ester mixture afforded the bromofuranones (3)-(5) [20] (Figure 4); and third, the reductive dehalogenation with zinc of (3) afforded (1) in 10% overall yield (Figure 5). Structure identity of (1) was also established by comparison of their 1H and 13C NMR spectra with data reported in the literature.
Protoanemonin (5-methylene-2(5//)-furanone) (1). Amber oil, bp 57-59 °C (lit 58-60 °C [15,17]), UV(H2O) λ max (log ε) 260 nm , H NMR (CD3OD, 400 MHz) δ: 6.3 (1H, ddd, .7=5.5, 1.8, 0.9 Hz, H-3), 7.68 (1H, dt, 7=5.5, 0.5 Hz, H-4), 5.23 (1H, ddd, 7=2.4, 1.8, 0.5 Hz, H-6Z), 5.03 (1H, ddd, 7=2.5, 0.9, 0.5 Hz, H-6E); 13C NMR (CD3OD, 400 MHz, assignment by DEPT and HSQC) δ: 170.4 (C-2), 121.0 (C-3), 143.9 (C-4), 155.5 (C-5), 97.0 (5-CH2); EI MS m/z (rel. int.): 96 [M+] (100), 68 (36), 54 (12), 42 (28), 40 (8), 39 (4).
The reductive dehalogenation reaction with zinc was a critical step of the synthesis. Zinc was considered for this step since its efficiency as a reductive agent, and it exerts lower toxicity compared to other metals. The halogen/hydrogen exchange from (3) to (1) could progress by the well-known radical mechanism [21,24], that should proceed through the next steps: first, the haloalkene captures an electron from the metal to give an anion-radical; subsequently, the fragmentation of the latter in a radical and an halide ion occurs, after which the yielded radical is reduced to an anion; and finally, the anion protonation to generate the dehydrohalogenated products takes place. In this way, the method employed in this study provides a simple and affordable synthesis of protoanemonin.
Biological activity
The cytotoxic activity of protoanemonin (1) and furanones (3)-(5) was evaluated against two human cancer cell lines: PC-3 (prostate adenocarcinoma) and U-251(glioblastoma). The evaluation was carried on by SRB assay and using cisplatin as the positive control (Table 1).
Table 1 Evaluation of cell growth inhibitory (IC50 ± SD) activity against PC-3 and U-251 cell lines.
Previous reports had described weak and moderated activities in vitro of protoanemonin isolated from Clematis herb in four cancer cell lines [14]. Nevertheless, our investigation found a strong cytotoxic activity of synthesized protoanemonin (1) and brominated furanone derivatives (3), (4) and (5) in PC-3 and U-251 human cell lines. The IC50 values were in the range of 0.31 to 7.30 µM. These compounds were also more active than the reference drug cisplatin. Worth mentioning that keto methyl ester 2c, precursor molecule of most active brominated furanone (5), was also screened for PC-3 and U-251 growth inhibition in 50 µg/mL concentration, with poor results: about 15% and 5% inhibition, respectively. Hence this indicates that 5-methylene-2-(577)-furanone ring is essential for the biological activity. Both in PC-3 and U-251 cell lines the sequence from the highest to the lowest cytotoxic activity of furanones was (5)>(4)>(1)>(3)>cisplatin. It seems that the presence of a bromine atom in the five-membered ring reduces cytotoxicity as in (3). Protoanemonin, (1), does not have bromine atoms but exhibited a strong cytotoxic effect and was more active than (3), which showed less activity than (4) and (5). Therefore, 5-methylene-2-(5i7)-furanone ring is necessary to exert the biological activity, but bromine atoms presence in 1' position is necessary to increase the cytotoxicity as in furanone (5). Bromine substituents are widely used in drug-like compounds as hydrophobic moieties and participating as Lewis bases (electron donor); but halogen substituents can also act as Lewis acids by forming directed close contacts with oxygen, nitrogen, or sulfur with other molecules [25]. In vitro assays of a related molecule 5-(bromomethyl)furan-2(577)-showed to promote apoptosis and cell cycle arrest in HepG2 (liver) and A549 (lung) human neoplastic cells, mediated by mitochondrial extrinsic, or death receptor pathways [14]. This biological action could be shared by the furanones here studied. However, the protoanemonin and bromo furanones (3) -(5) mechanisms of action against cancer cell lines must be explored.
These results suggest that position and type of substituent in 5-methylene-2-(5H)-furanone is crucial for biological activity as (1), (3), (4) , and (5) showed potent activity against cancer cells. While it has been reported conversely that methyl protoanemonin (methyl residue at 1' site) acts as a weak tumour inducer [26].
Considering the concept of the threshold of toxicological concern (TTC) and based on structural alerts, protoanemonin has been classified as a Cramer class III compound a TTC of 180 µg/day in adults [26]. Notwithstanding, the significant antiproliferative effect of (1) supports protoanemonin ethnomedical use as anticancer agents of Ranunculaceae plants. This is the first time that brominated derivatives of protoanemonin are studied against cancer cell lines and could be considered as potential anticancer agents, according to the cytotoxic assay. Protoanemonin (1) and brominated derivatives may be a lead for further studies on its mechanism of action in cancer cells.
Cytotoxic dibromo furanone (3) (C30), has been intensively used as a quorum sensing inhibitor in bacterial studies. The results of this study imply the feasibility of (3) to be included in the group of anticancer drugs that have been recently used against resistant bacteria [27].
Conclusions
A concise protoanemonin synthesis was achieved by using a reductive dehalogenation step mediated by zinc. It was found that protoanemonin (1) and the brominated furanones (3)-(5) possess a potent antiproliferative activity. The bioactivity is enhanced with one bromine exocyclic substitution. It is worth noting that (3) is a remarkable inhibitor of bacterial quorum sensing and possesses a potent cytotoxic activity against human cancer cell lines. The current findings could be adequate for the development of antitumor 5-methylene-2(5H)-furanone derivatives.

