











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
Myotonic dystrophy type 1 (MD1), commonly referred to as Steinert disease, is a multisystem condition mostly affecting the skeletal muscle, the central nervous system, the respiratory, gastrointestinal, cardiac and endocrine systems in neonates, children, and adults. MD1 is an inherited autosomal dominant pathology with variable penetrance that develops as a result of an expanded CTG triplet repeat of a noncoding DNA segment on the DMPK gene in chromosome 19ql3.3. The incidence and the prevalence of MD1 are approximately 13/10,000 and 2 to 14/100,000, respectively. These patients are at a higher anesthetic risk mainly due to the muscular dysfunction that further deteriorates the respiratory function and to the multiple organ involvement.1,2
Clinical case
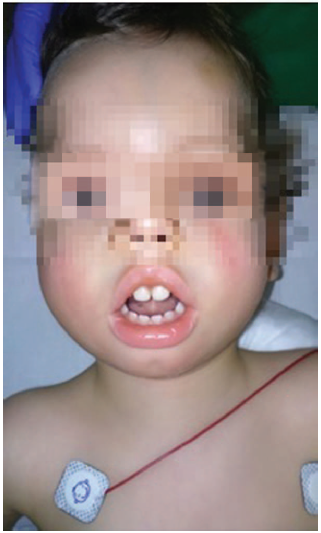
This is a 22-month-old male patient with 9kg of weight who underwent a programmed bilateral orchiopexy. The patient has a history of polyhydramnios during pregnancy and was born through cesarean section due to risk of fetal discomfort at week 41+3. The Apgar score was 1/10 associated with meconium aspiration that required orotracheal intubation and postnatal intensive care unit (ICU) monitoring. At birth, the patient was diagnosed with MD1, as well as the mother who did not show any evidence of myotony. The patient presents with inexpressive facies, fish-like mouth, retrognathia and prominent forehead, typical of MD 1 (Fig. 1), hypotony, bilateral pyelectasis, reducible equinus feet, and episodes of atrial extrasystole with aberrant conduction. The usual treatment in based on budesonide, montelukast, flecainide, and atenolol.
In the operating room (OR), the patient was under noninvasive blood pressure, ECG, pulse oximetry, and esophageal temperature monitoring. Inhaled anesthesia induction was administered with sevofluorane and #1.5 laryngeal mask was placed to start assisted ventilation with support pressure and expired CO2 monitoring. The halogenated agent was maintained with a 2.8% minimum alveolar concentration throughout the procedure. Pain was controlled with a caudal puncture and the administration of a (9 ml) bupivacaine bolus 0.125% and lidocaine 0.5%, with ultrasound-guided catheter insertion up to the T12 level, followed by ropivacaine 0.1% perfusion. During the intervention, a continuous perfusion of remifentanil 0.1 mg/kg/min was administered. The intervention was uneventful and the patient woke up in the OR, and was then transferred to the pediatric ICU for monitoring during the first 24hours, maintaining continuous ropivacaine 0.1% perfusion via caudal catheter for postoperative pain control. No complications developed.
Discussion
The anesthetic management of these patients needs to take several special considerations into account, in addition to avoiding any potential triggers of a myotonic crisis.
Premedication with benzodiazepines is contraindicated in these patients because of their muscular and respiratory disease; due to the high risk of respiratory depression, they come to the OR with no premedication.1,2
Halogenated anesthetic agents may be safely used for induction and maintenance of anesthesia; although the coding gene is located in the same chromosome as for malignant hyperthermia, the use of this type of anesthetic agents has not been associated with the development of this severe condition3 in these patients. Furthermore, in case of a myotonic crisis, halogenated compounds help to alleviate the crisis, while depolarizing relaxants are useless, as the source of the contraction is the membrane hyperexcitability.
The use of opioids for pain control may further intensify any underlying respiratory problems; however, on the basis of its short half-life, remifentanil is a good option to control acute postoperative pain. Regional anesthesia has been successfully reported in the literature to reduce the administration of opioids for managing intraoperative and postoperative pain.1-3
Hypothermia must be prevented, as it may trigger a myotonic crisis and this is achieved by maintaining adequate room temperatures, in addition to the use of a thermal blanket and fluid warming as needed.1,3
Succinylcholine is contraindicated, as it may trigger dose-dependent myotonies; the use of short-acting non-depolarizing muscle relaxants is allowed, as long as the dose is adjusted and the effect is monitored.1,2
With regards to myotonic dystrophy type 2, or proximal myopathy, the considerations for managing anesthesia are similar, though these are patients who may be asymptomatic until adulthood and represent a lower anesthetic risk.2
Finally, each patient must be managed in an individualized manner and this case just emphasizes the importance of a comprehensive pre-anesthesia evaluation, careful intraoperative management avoiding hypothermia, stress, and pain when volatile anesthetic agents, nondepolarizing muscle relaxants, short half-life opioids, and locoregional anesthesia are allowed, in addition to an intensive postoperative vigilance.
Ethical disclosures
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of data. The authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consent. The authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.

