











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introducción
La enfermedad de Pompe (EP) es una enfermedad por depósito lisosomal que se caracteriza por la deficiencia de la enzima alfa-glucosidasa ácida, lo que causa un acumulo anormal de glucógeno en el músculo y en otros tejidos. Esto último, a su vez, genera debilidad muscular progresiva, alteración respiratoria, trastorno de la funcionalidad motora y muerte prematura, cuya gravedad y velocidad de deterioro dependerán del porcentaje de enzima residual 1. La instauración de un tratamiento multidisciplinario y modificador de la enfermedad puede lograr no solo mejorar la sobrevida del paciente, sino su funcionalidad, en especial cuando se inicia de forma oportuna y se utilizan estrategias tanto farmacológicas como no farmacológicas. En este artículo nos centramos en revisar las características de un adecuado tratamiento nutricional y gastrointestinal, así como el uso y la aplicación de la terapia de reemplazo enzimático.
Evaluación y tratamiento nutricional, alimentario y gastrointestinal
Los pacientes con enfermedad de Pompe de inicio tardío (LOPD) cursan con dificultad para alimentarse y tragar, los factores descritos que contribuyen a esta circunstancia son la debilidad de los músculos bulbares, la hipotonía facial, la fatiga de los músculos faciales, la debilidad de la lengua, la macroglosia, la fatiga, el reducido rango de movimiento de los músculos de la masticación y la alteración de los movimientos orales 1. La dificultad para alimentarse y tragar produce una ingesta inadecuada de proteínas y como resultado la degradación endógena de estas. Adicionalmente, la desnutrición puede agravar la debilidad muscular 2.
La debilidad lingual significativa, la disfagia orofaríngea y los diferentes grados de disfagia aumentan el riesgo de trastornos secundarios como microaspiraciones y neumopatías 3. Asimismo, los pacientes tienen dificultad para el control de la saliva y las secreciones, que tienden a acumularse en las cuerdas vocales, lo cual puede desencadenar complicaciones respiratorias 3.
Para asegurar un adecuado soporte nutricional, los pacientes con EP requieren una evaluación nutricional integral desde el diagnóstico y controles periódicos durante la evolución de la enfermedad. El soporte nutricional y alimentario debe estar a cargo de profesionales de la salud especialistas en el tema, un nutricionista experimentado, de preferencia uno que se especialice en trastornos metabólicos, quien desarrollará un plan de alimentación personalizado, teniendo en cuenta la tolerancia y las dificultades en la masticación y la deglución 4.
Se deberá realizar un monitoreo, un seguimiento y una educación regular de los hábitos alimentarios, considerando la cantidad, el tipo de alimentos ingeridos, las intolerancias y las dificultades del paciente y su familia. Es importante capacitar o informar al paciente y a su familia sobre la elección, la combinación y la preparación de los alimentos para favorecer la adherencia al tratamiento.
Hasta la fecha no hay datos suficientes para recomendar algún tipo de régimen dietético específico 5,6, sin embargo, una dieta alta en proteínas y baja en carbohidratos mostró una mejoría en la fuerza muscular en estos pacientes 7. Otros autores sugieren que los pacientes se podrían beneficiar de una disminución del contenido de grasas y carbohidratos, especialmente los refinados, y del aumento de aminoácidos 6-8. Se debe considerar el aumento de la ingesta de la fibra alimentaria (soluble e insoluble) y el uso de suplementos dietarios 6.
Una dieta alta en proteínas (20-25 % de la ingesta energética total de proteínas, 30-35 % de carbohidratos y 35-40 % de lípidos), con especial atención a las vitaminas y los minerales, puede beneficiar al paciente. El suplemento con 1,4 g de L-Alanina hasta 4 veces al día, está indicado si no se puede alcanzar el contenido de proteína recomendado, asociado con un 4 % (500 ml) de aminoácidos de cadena ramificada 4.
Se han descrito síntomas gastrointestinales como diarrea crónica, estreñimiento, molestias o calambres abdominales, plenitud postprandial o saciedad temprana, y pobre ganancia de peso en la adolescencia 9. La disfunción digestiva ha sido reportada en la literatura, así como su mejoría con la terapia de reemplazo enzimático (TRE) 2. Adicionalmente, se sugiere que la TRE puede mejorar los síntomas gastrointestinales en pacientes con enfermedad de Pompe de inicio tardío; no obstante, se requieren más estudios al respecto 10.
En casos de enfermedad avanzada, podría ser necesario proteger al paciente del riesgo de broncoaspiración, suspendiendo la alimentación por vía oral y mediante el uso de recursos como la sonda nasogástrica, la gastrostomía o la gastroyeyunostomía 11.
Siempre se debe realizar una evaluación por fonoaudiólogo y videofluoroscopía.
El estado basal nutricional es un factor pronóstico de los resultados de la TRE, el IMC (índice de masa corporal) recomendado para estos pacientes es el mayor o igual a 18 kg/m y debe ser tomado en cuenta para el inicio del tratamiento con TRE 10-12.
Terapia de reemplazo enzimático
Generalidades
La terapia de sustitución o reemplazo con la enzima α-glucosidasa recombinante humana (rhGAA) alglucosidasa, aprobada para su uso en los Estados Unidos y Europa desde el 2006, y disponible en Colombia desde el 2015, es por el momento el único tratamiento modificador de enfermedad avalado en Colombia, el cual se administra en infusión endovenosa a una dosis de 20 mg/kg cada 2 semanas 13. Existen limitantes farmacológicas como la eliminación no productiva o "efecto sifón" (gran parte de la rhGAA es capturada por lo lisosomas de células no musculares) y la limitada internalización de la rhGAA en los miocitos debido a la escasa fosforilación de manosa 6-fosfato, lo cual genera que solo el 1 % de enzima eficaz llegue a los lisosomas deseados 1. Por otra parte, debido a que la alglucosidasa no es la enzima natural del paciente, se puede activar una respuesta inmunológica de anticuerpos relacionada con el material inmunológico de reacción cruzada (CRIM), en especial cuando este es negativo (ausencia de GAA natural), como sucede en la enfermedad de Pompe de inicio infantil (IOPD), en la cual en algunos casos esta producción de autoanticuerpos puede neutralizar el efecto de la terapia de reemplazo enzimático (TRE) 14. A pesar de estas limitantes, la TRE con rhGAA ha cambiado el curso de la enfermedad y la calidad de vida de los pacientes, en especial cuando se inicia de forma oportuna.
Evidencia de la eficacia y la seguridad
Los estudios multicéntricos iniciales, incluidos los pivotales patrocinados por la industria, confirmaron la eficacia y la seguridad del uso de la TRE en pacientes con EP infantil menores de 6 meses, en quienes se demostró una dramática mejoría en la sobrevida (99 % a los 18 meses y 67,5 % a los 3 años), un 50 % menos de requerimiento de ventilación mecánica y una independencia en la marcha en el 40 % de los pacientes 15. También se demostró una mejoría de la cardiomiopatía, una reducción de la mortalidad en un 79 % y un decremento en el uso de ventilación mecánica en un 58 % 16,17. A pesar de esto, no todos los pacientes alcanzan una independencia motriz, y si bien la función respiratoria y la deglutoria contribuyen a mejorar la calidad de vida, esta permanece afectada 18.
Posteriormente, el estudio LOTS (Late Onset Treatment Study) demostró en pacientes con EP tardía que el uso de la TRE lograba una mejoría en la fuerza muscular, medida por la prueba de caminata de 6 minutos (6MWT), y una estabilidad de la capacidad vital forzada a 2 años de seguimiento 19.
Una revisión sistemática encontró que en pacientes con EP tardía se logró mejorar, en el primer año de TRE, un 78 % la distancia recorrida en el 6MWT; un 10 % de los pacientes postrados en silla de ruedas logró mejoría de su independencia; el 53 % mejoró su capacidad vital forzada (CVF) y un 70 % de los pacientes con ventilación invasiva mejoraron sus parámetros respiratorios 20.
Recientemente, en otra revisión sistemática, esta vez con metaanálisis, se demostró una mejoría significativa en la distancia recorrida medida por 6MWT, pero no se encontraron cambios en la CVF, aunque hubo una estabilización o al menos una desaceleración del decremento 21.
Finalmente, en el estudio STIG (Spain, Taiwan, Italy, Germany), con un seguimiento a más de 10 años, se demostró en pacientes con variantes tardías de la EP, estabilidad o mejoría de la fuerza muscular y una mejoría inicial del 33 % de la capacidad vital forzada (CVF), con posterior disminución en la velocidad de deterioro, en comparación con la esperada para la enfermedad (1-4,6 % anual) 22.
En cuanto a la seguridad de la TRE, la frecuencia de efectos adversos graves es rara y está relacionada en su mayoría con la respuesta de hipersensibilidad mediada por inmunoglobulina E, la cual puede ser manejada con terapia de desensibilización de la rhGAA o inmunomoduladores. Por su parte, los efectos adversos leves a moderados tienden a presentarse más frecuentemente, y en su mayoría se relacionan con reacciones asociadas a la infusión, fácilmente manejables con premedicación, tratamiento sintomático o cambios transitorios de la tasa de infusión 23.
Valoración pretratamiento
Una vez obtenido el diagnóstico patológico y molecular, es determinante elaborar el perfil inicial del paciente con EP, no solo porque de esta información depende la indicación de iniciar tratamiento, sino porque permite establecer los parámetros basales, los cuales deberán ser seguidos para evaluar la respuesta al tratamiento (tabla 1) 1.
Tabla 1 Evaluación pretratamiento de TRE
| Aspecto | Análisis |
|---|---|
| Laboratorio | CPK, AST, ALT, LDH, GGT, Creatinina sérica, BUN, hemograma y glucosa |
| Musculoesquelético | Exploración de fuerza, tono y trofismo muscular completa, y uso de pruebas funcionales según el estado clínico del paciente (e. g., 6MWT) y EMG/NC (complemento a exploración física). Opcional: resonancia de músculo (según criterio del especialista)* |
| Cardiovascular | Electrocardiograma y ecocardiograma. Resonancia cardíaca (opcional y con base en criterio del especialista) |
| Respiratorio | Espirometría con broncodilatador, polisomnografía con oximetría, radiografía de tórax, gasometría arterial (en EP con insuficiencia respiratoria). En caso de pacientes con uso de ventilación no invasiva o invasiva, se podrán requerir otros parámetros (según criterio de especialista) |
| Nutricional | Peso, talla, IMC, perímetro cintura/cadera, pliegue tricipital (niños), perímetro braquial (niños), densitometría ósea y proteínas totales/fraccionadas |
| Neuropsicológica | Exploración neurológica completa, valoración psicológica y de red de apoyo social. Audiometría (opcional) |
| Inmunológico | Evaluación de CRIM en IOPD o según criterio del médico tratante |
| Fonoaudiológica | Exploración física fonoaudiológica, deglución, funcionalidad orofaríngea y evaluación de la audición |
*Se puede realizar de forma segmentaria o corporal total, y con técnica semicuantitativa o cuantitativa.
CPK: creatin kinasa, AST: aspartato aminotransferasa, ALT: alanina aminotransferasa, LDH: lactato deshidrogenasa, GGT: gamma glutamil transferasa, BUN: nitrógeno ureico, EMG/NC: electromiografía y neuroconducciones, IMC: índice de masa corporal, CRIM: material inmunológico de reacción cruzada
Fuente: 1
Indicaciones para el inicio de la terapia de reemplazo enzimático
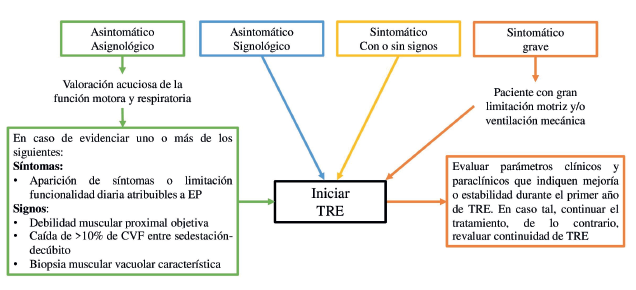
Debido a la heterogeneidad clínica en pacientes con EP, la variabilidad intrafamiliar, los diferentes genotipos (variantes patológicas, indeterminadas, pseudodeficiencias, polimorfismos, etc.) y la posibilidad de tener pacientes portadores asintomáticos y asignológicos, o con ausencia de signos objetivo, hacen que para determinar el inicio de la TRE no solo se tenga en cuenta el análisis molecular de la mutación, sino su correlación clínica y la demostración de la baja actividad enzimática en sangre o fibroblastos, ya que no todos los pacientes que tengan la variante patogénica tienen asimismo la enfermedad, y por tanto no son candidatos a recibir TRE (figura 1) 1,2.
Metas de la terapia de reemplazo enzimático
Después de tomar la decisión de iniciar la TRE, y tras una evaluación del perfil del paciente, se deben establecer cuáles serán las metas del tratamiento, las cuales siempre se deben fijar de forma individualizada. Dentro de los objetivos de la TRE se encuentran los siguientes 2:
a) Retrasar, estabilizar o mejorar la progresión de la patología.
b) Reducir las comorbilidades e incrementar la sobrevida.
c) Optimizar la fuerza muscular, promover la movilidad, mejorar el condicionamiento físico y retrasar la necesidad de uso de dispositivos para la marcha.
d) Mejorar o preservar la independencia funcional del paciente y mejorar la calidad de vida.
e) Mantener o retrasar el deterioro de la función respiratoria.
f) Mejorar los parámetros ventilatorios invasivos o no invasivos.
Seguimiento del tratamiento
Una vez establecido el perfil inicial del paciente y las metas del tratamiento, es fundamental hacer un seguimiento suficiente y eficiente, no solo para definir la continuidad de la TRE, sino para ajustar el tratamiento multidisciplinario, en especial los programas de rehabilitación física, ocupacional, fonoaudiológica, respiratoria y nutricional. La valoración física es prioritaria y se debe hacer por medio de pruebas de evaluación de fuerza muscular, ya sean manuales, como el Manual Muscle Testing (MMT), o por dinamometría con el Quantitative Measure Test (QMT) o el Hand Held Dynamometer (HHD). Del mismo modo, es necesario llevar a cabo pruebas de valoración funcional cronometradas, como el signo de Gowers (pararse desde una posición de decúbito supino), la prueba de los cuatro escalones, la prueba de correr y caminar en 10 metros, la 6MWT y la prueba de levantarse desde una silla con y sin apoyo 1,2. El esquema que se presenta en la tabla 2, es una guía general, y puede ser insuficiente para el seguimiento de pacientes pluripatológicos, con ventilación invasiva o con patología neuromuscular avanzada 24.
Tabla 2 Seguimiento del tratamiento con TRE
| Frecuencia de seguimiento o evaluación en meses | |||
|---|---|---|---|
| Aspecto | IOPD | LOPD | EP bajo ventilación |
| Evaluación motora+ | 3 | 6 | 3 |
| Evaluación deglutoria | 3 | 6 | 3 |
| Valoración nutricional~ | 3 - 6 | 6 | 6 |
| Evaluación respiratoria | 3 | 6 | 3 |
| Valoración médica | 1 | 3 | 1-3 |
| Evaluación de calidad de vida* y cuidadores | 6 | 6 | 6 |
| Anticuerpos IgG anti rhGAA | 3 (primeros 2 años) luego anual | SC | SC |
| Ecocardiograma | 3 | SC | SC |
| ECG / holter | 3 | 12 | 6-12 |
| Espirometría | 1 | 3 | 1-3 |
| Polisomnografía | SC | 6 | 3 |
| Radiografía de tórax | 3 | 6-12 | 3-6 |
| Resonancia muscular^ | 12-18 | 12-18 | 12-18 |
| Gasometría arterial | 3 | SC | 3-6 |
| Densitometría ósea. | SC | SC | SC |
+ Incluir pruebas de valoración de fuerza muscular y funcional cronometradas, así como exploración de funcionalidad instrumental y de actividades básicas.
~Incluir valoración de síntomas gastrointestinales.
*Usar escalas como el SF-36.
^Correlación clínico-patológica ya sea por técnica semicuantitativa (T1 = infiltración grasa y T2/STIR = edema/inflamación) o cuantitativa (porcentaje de cambio de fibra muscular) (referencia).
VPPNI: ventilación con presión positiva no invasiva, VI: ventilación invasiva, ECG: electrocardiograma, DEXA: densitometría ósea, SC: según criterio clínico.
Suspensión de la terapia de reemplazo enzimático
¿Qué sucede al dejar de infundir?
Descontinuar o suspender la TRE, incluso por periodos cortos (3 meses en adelante), implica una progresión de la patología con decremento de la CVF y de la distancia recorrida medida por 6MWT, además, este daño puede ser irreversible, ya que a pesar de reiniciar la TRE, puede no lograrse la recuperación de la lesión muscular generada durante el periodo de suspensión 25. Por consiguiente, la recomendación es mantener la TRE cada 2 semanas de forma estricta, y evitar interrupciones que puedan impactar negativamente en la calidad de vida del paciente.
¿Cuándo dejar de infundir?
La decisión de suspender una TRE se debe tomar de forma objetiva y consensuada (especialistas tratantes y deseo del paciente), siempre valorando el balance riesgo-beneficio de su interrupción. Una de las herramientas más utilizadas son los criterios EPOC (European Pompe Consortium), los cuales determinan 6 escenarios de suspensión de TRE: 1) reacciones adversas relacionadas con la infusión que no pueden ser manejas apropiadamente; 2) títulos elevados de anticuerpos IgG neutralizantes del efecto de la rhGAA; 3) deseo del paciente de no continuar con la terapia; 4) mala adherencia del paciente (falla al cumplimiento de las infusiones y del seguimiento clínico); 5) presencia de otra patología avanzada, en estadio terminal o donde es inapropiado sostener la vida; 6) ausencia de mejoría o estabilización de los parámetros musculares o respiratorios durante los primeros 2 años de inicio de la TRE (o durante el primer año en caso de pacientes con EP sintomáticos graves) 26.
Tratamiento de reemplazo enzimático en enfermedad de Pompe en población pediátrica
La TRE con rhGAA fue aprobada como el primer tratamiento para la enfermedad de pompe por la Food and Drug Administration (FDA) en el 2006 27-29. En los pacientes con enfermedad de Pompe infantil clásica (IOPD), la terapia de reemplazo enzimático ha demostrado mejorar la sobrevida, hipertrofia cardíaca y adquisición de hitos del desarrollo motor, especialmente cuando se inicia antes de los 6 meses de edad. Sin embargo, la mortalidad sigue siendo elevada (25-43 %) 27-29. Los factores predictores de mal pronóstico en TRE incluyen un inicio tardío en relación con el tiempo de evolución de las manifestaciones clínicas, ausencia completa de actividad enzimática nativa o estatus de material inmunológico cruzado negativo (estatus CRIM negativo), sexo masculino, debilidad muscular severa y una duración prolongada de la enfermedad previamente al inicio del tratamiento farmacológico 30. Por el contrario, los predictores de una buena respuesta farmacológica incluyen un inicio temprano de la terapia y bajos títulos de anticuerpos anti-rhGAA 15.
La relación entre el estatus CRIM y la respuesta a la terapia de reemplazo enzimático es explicada por la posibilidad de desarrollar autoanticuerpos que neutralicen su efecto. Si bien es cierto que el 89 % de los pacientes con enfermedad de Pompe infantil, que por regla general tienen una actividad enzimática nativa inferior al 1 % o nula, los desarrollarán, la mayoría de ellos generará tolerancia inmunológica al continuar el tratamiento. Solamente aquellos con títulos de anticuerpos altos y sostenidos (≥ 1:51,200 en dos ocasiones ≥ 6 meses) contra la alfa glucosidasa recombinante humana (IgG anti-rhGAA) tendrán un mal pronóstico, con una edad promedio de muerte de 27,1 meses (29,30). A diferencia de lo anterior, los pacientes con preservación de algún grado de actividad nativa de la alfa glucosidasa (estatus CRIM positivo), generalmente no desarrollan o presentan o bajos títulos de IgG anti-rhGAA, y mantienen una mejor respuesta en el tiempo a la terapia de reemplazo enzimático, lo cual se evidencia clínicamente por una menor mortalidad y un menor requerimiento de ventilación mecánica invasiva en relación con los pacientes CRIM negativos 31.
La TRE está indicada en todos los casos de enfermedad de Pompe infantil tan pronto como se confirme el diagnóstico, y debe establecerse además el estatus CRIM del paciente e iniciar una terapia de tolerancia inmunológica en los casos CRIM negativos, con fármacos que actúen o eliminen la proliferación de las células B y T, como el rituximab, el metrotexato, la metilprednisolona o la inmuno-globulina en mono o politerapia 32,33. La terapia de reemplazo enzimático se realiza a una dosis estándar de 20 mg/kg/dosis cada dos semanas 33, algunos estudios sugieren que dosis mayores de 40 mg/kg podrían llegar a mejorar el pronóstico clínico, pero la evidencia es insuficiente 34. Se recomienda una velocidad de infusión de 1 mg/kg/h (30 minutos), 3 mg/kg/h (30 m), 5 mg/kg/h (30 m) y 7 mg /kg/h (hasta finalizar la infusión), con una duración promedio de 3 horas y 45 minutos; esta velocidad puede ser prolongada en caso de rash o alergia 34.
La investigación inicial con la cual fue aprobada la TRE en el 2006 con rhGAA derivada de células de ovario de hámster chino, fue un estudio de tipo abierto multicéntrico que incluyó a 8 pacientes con IOPD y evaluó la eficacia y la seguridad del fármaco a 52 semanas. Este estudio demostró una mejoría en la cardiomiopatía hipertrófica en los 8 participantes, 6 pacientes lograron sobrevivir, 5 estuvieron libres de ventilación invasiva, 3 lograron marcha independiente (inicio de la terapia antes de los 6 meses), y se modificó la historia natural previamente descrita de la enfermedad 35. Un segundo ensayo clínico evaluó la eficacia de la TRE en 18 pacientes con IOPD < 7 meses vs. 168 controles históricos, para lo cual se hizo una aleatorización a la dosis de 20 mg/kg/dosis o 40 mg/kg/dosis. El estudio demostró una reducción del riesgo de muerte del 99 % (hazard ratio 0,01, IC 95 % 0,00-0,10, p = 0,001), una disminución del riesgo de muerte o ventilación invasiva del 92 % (hazard ratio 0,08, IC 95 %: 0,03-0,21, p = 0,001), y una reducción del riesgo de muerte o cualquier tipo de ventilación de un 88 % (hazardratio 0,12, IC 95 %: 0,05-0,29, p = 0,001). Adicionalmente, a la semana 52 el índice de masa del ventrículo izquierdo disminuyó a 86,8 g/m2, a partir de un índice basal de 193,4 g/m2. Lo anterior, tan tempranamente como a las 4 semanas de tratamiento farmacológico 36. Múltiples estudios posteriores han corroborado la eficacia del fármaco en el tratamiento de los pacientes con enfermedad de Pompe infantil, incluidos estudios de extensión que han demostrado la eficacia del fármaco a largo plazo, principalmente en términos de mortalidad, si bien no se logra una estabilización completa de la patología ni evitar en la mayoría de los casos el requerimiento de ventilación invasiva 12,37,38.
Condiciones especiales
Anestesia / cirugía
Debido al incremento de complicaciones durante procedimientos quirúrgicos en los pacientes con patología neuromuscular, en especial por el uso de ha-lotano y relajantes musculares, se hacen las siguientes recomendaciones 2:
Realizar procedimientos anestésicos solo cuando sea necesario.
En caso de requerir varios procedimientos quirúrgicos, de ser posible, unificarlos en un solo momento anestésico.
Evitar intubación endotraqueal. En caso de ser necesario, realizarlo solo por personal experto.
Realizar una revisión intraoperatoria rigurosa.
Usar anestésicos inhalados solo en pacientes que no sean sintomáticos graves.
Evitar agentes despolarizantes, por el riesgo de hiperpotasemia.
Monitorizar el volumen circulante efectivo.
De ser posible, realizar procedimientos quirúrgicos en centros que cuenten con personal con experiencia en el tratamiento de EP.
Lactancia / embarazo
La evidencia científica es precaria en estas condiciones, puesto que en ningún estudio pivotal fueron incluidas mujeres en embarazo o lactancia, sin embargo, desde la aprobación del tratamiento, inicialmente se consideró categoría B de riesgo obstétrico, pero recientemente fue recategorizado a C debido a que en modelos animales se había demostrado cierta teratogenicidad, en ausencia de estudios controlados en mujeres 2. Desde el 2014 se han reportado múltiples series de casos de pacientes embarazadas y uso de TRE, con desenlaces favorables, sin evidencia de malformaciones fetales ni complicaciones adicionales (solo las esperadas en una enfermedad neuromuscular) durante el parto y el postparto, y en cambio se encontró una progresión de la patología en mujeres embarazadas que no continuaban con la TRE 39. Por tal motivo, se sugiere evaluar el perfil basal de la patología (CVF, cardiovascular y muscular), para así explicar a la paciente los riesgos potenciales para ella y para el feto en caso de usar o no TRE durante todas las fases del embarazo, y se recomienda el parto por cesárea con anestesia local para evitar complicaciones 2.
Reacciones asociadas a la terapia de reemplazo enzimático y su tratamiento
La TRE con rhGAA puede generar dos tipos de respuestas inmunológicas: la primera es la generación de anticuerpos IgG específicos contra la terapia de reemplazo enzimático, que pueden ser de tipo neutralizante -cuando estos anticuerpos se unen a dominios funcionales, de entrada o catalíticos de la rhGAA- o no neutralizante -cuando se unen a receptores Fc de macrófagos o monocitos- 40. En paciente con Pompe de inicio infantil, sería idóneo conocer el estado CRIM previo a la TRE, ya que en pacientes CRIM negativo se debe iniciar una terapia de inmunomodulación antes del inicio de TRE con rituximab (375mg/m2), cada semana por 4 semanas, y luego de mantenimiento, según parámetros linfocitarios, metotrexate (0,4 mg/kg) oral cada semana e inmunoglobulina G endovenosa 400-500 mg/kg cada 4 semanas, con medición permanente de tirulos de anticuerpos IgG específicos contra rhGAA 41. En caso de no poder obtener el estado CRIM, o si este es dudoso, se recomienda manejar a los pacientes con Pompe infantil como si fuesen CRIM negativos, para no retrasar el inicio de la TRE 40.
El segundo tipo de reacción es la hipersensibilidad asociada a la rhGAA, la cual a su vez puede ser de tipo alérgico, mediada por antígeno específicos (asociada o no a IgE), o de tipo no alérgico, mediada por otros mecanismos como la activación del complemento, laq activación directa de mastocitos, o la liberación de citocinas, entre otros 40. El 30-40 % de las reacciones de hipersensibilidad son leves y se consigue solventarlas con el tratamiento estándar que incluye premedicación con analgésicos y antihistamínicos, así como cambios en la rata de infusión de la TRE 40. El 14 % de los pacientes puede presentar reacciones moderadas, con compromiso de al menos 2 sistemas (cardiovascular, respiratorio, piel, nervioso), y en el 1 % se puede observar choque anafiláctico 40. En caso de que el tratamiento estándar no mejore las reacciones de hipersensibilidad asociadas a la TRE, o que exista una reacción moderada a grave, se debe considerar utilizar la terapia de desensibilización, dependiendo del perfil de cada paciente y con apoyo del servicio de alergología, con dosis de TRE de 1/1.000.000 - 1/100 de la dosis total, e incrementos de infusión paulatinos cada 15-20 minutos del doble o del triple de la dosis inmediatamente anterior, con el fin de modular el sistema inmunológico y permitir una mejor tolerancia, para así evitar suspender la TRE 40.
Tratamientos futuros
Debido a las limitaciones que aún ofrece la TRE con alglucosidasa, en la actualidad se investigan otros tratamientos con diferentes blancos de acción lisosomal que permitan mejorar aún más la evolución de la EP. La segunda generación de rhGAA, llamada avalglucosidasa, es una versión mejorada de la al-glucosidasa debido a su mayor carga de manosa-6 fosfato que garantiza una mayor internalización celular de la rhGAA. Esta se administra a una dosis de 40mg/kg cada 2 semanas (el doble de la alglucosidasa) y ha demostrado de forma contundente una mayor eficiencia terapéutica reflejada en los resultados del estudio COMET, cabeza a cabeza con la alglucosidasa, donde se observa superioridad en la respuesta de recuperación de la CVF y no inferioridad en el test de marcha de los 6 minutos (6MWT), así como mayor seguridad debido a una menor generación de autoanticuerpos y menores efectos asociados a infusión, por lo cual ha sido aprobada en varios países, incluidos algunos de Latinoamérica 42,43. En estudio fase 3 en curso se está investigando el uso combinado de TRE y chaperona, la cual permite estabilizar (doblaje apropiado) la enzima activa, para así restaurarla y potenciarla. También en fase 1 y 2 se encuentra el uso de terapia génica, especialmente mediante el uso de un vector viral para transportar la copia funcional del gen GAA, y de tal manera corregir el defecto genético y generar producción de enzima natural. Finalmente, se están evaluando otras terapias, como la estimulación de exocitosis de glucógeno mediante factores de transcripción y la terapia de reducción de sustrato por medio de molécula pequeña para disminuir la biosíntesis de glucógeno; en ambos casos se busca la disminución del depósito anormal de glucógeno 44.
Conclusiones y recomendaciones
La enfermedad de Pompe es una patología multisistémica, cuyo pronóstico dependerá del inicio oportuno del tratamiento, la implementación de pautas nutricionales adecuadas, el establecimiento de programas de rehabilitación física, el abordaje multidisciplinario y el seguimiento de los parámetros clínicos y paraclínicos para cada una de las variantes de la enfermedad y grupos etarios.

