










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957versão On-line ISSN 2500-7440
Rev. colomb. Gastroenterol. vol.37 no.4 Bogotá out./dez. 2022 Epub 12-Jul-2023
https://doi.org/10.22516/25007440.815
Reporte de caso
Síndrome de Peutz-Jeghers: reporte de caso
1
*
 http://orcid.org/0000-0003-0614-5932
http://orcid.org/0000-0003-0614-5932
2
http://orcid.org/0000-0002-2999-901X
1Clinical-surgical gastroenterologist, Hospital Regional Manuela Beltrán del Socorro, Hospital Universitario de Bucaramanga, Clínica Los Comuneros, Gastrocal, Floridablanca. Bucaramanga, Colombia.
2Internist, Universidad Nacional de Colombia. Bogota, Colombia.
3Internist, Universidad de Santander. Bucaramanga, Colombia.
4Internist, Universidad de Santander. Bucaramanga, Colombia.
El síndrome de Peutz-Jeghers es una enfermedad hereditaria, autosómica dominante, caracterizada por la presencia de múltiples pólipos gastrointestinales de tipo hamartomatoso y se asocia con hiperpigmentación mucocutánea.
A continuación, se reporta un caso de un paciente de 25 años con historia de hemicolectomía derecha por una intususcepción ileocolónica secundaria a un pólipo gigante en el íleon terminal. Se trata de un paciente que consultó por rectorragia, con evidencia en el examen físico de lesiones hipercromáticas color café oscuro en la mucosa yugal. Se realizó una colonoscopia total, en la que se observaron múltiples pólipos. Se practicó una mucosectomía endoscópica a algunos de ellos, histopatológicamente compatibles con pólipos hamartomatosos.
Palabras clave: Síndrome de Peutz-Jeghers; pólipos hamartomatosos; lesiones hipercromáticas
Peutz-Jeghers syndrome is an autosomal dominant hereditary disease characterized by multiple hamartomatous-type gastrointestinal polyps associated with mucocutaneous hyperpigmentation.
A case of a 25-year-old male patient with a history of right hemicolectomy due to ileocolonic intussusception secondary to a giant polyp in the terminal ileum is reported. This patient consulted for rectal bleeding, with evidence on physical examination of dark brown hyperchromatic lesions on the buccal mucosa. A total colonoscopy was performed, noting multiple polyps. Endoscopic mucosectomy was conducted on some of them, being histopathologically compatible with hamartomatous polyps.
Keywords: Peutz-Jeghers syndrome; hamartomatous polyps; hyperchromatic lesions
Introducción
El síndrome de Peutz-Jeghers es una poliposis hereditaria, autosómica dominante, poco común, asociada con una alta frecuencia a mutaciones del gen STK11/LKB1, localizado en el brazo corto del cromosoma 191,4. Se caracteriza por una pigmentación mucocutánea y la presencia de múltiples pólipos hamartomatosos, con un mayor riesgo de malignidad gastrointestinal. También se ha asociado con aumento del riesgo de cáncer ginecológico, testicular y tiroideo1.
La pigmentación mucocutánea se debe a la presencia de macrófagos cargados de melanina en la dermis y el aumento de melanocitos en la unión dermoepidérmica, presente en alrededor del 95% de los pacientes. Estas suelen aparecer en la infancia alrededor de la boca, las fosas nasales, la región perianal, los dedos y los artejos2,5.
Los pólipos de SPJ se localizan habitualmente a nivel del intestino delgado, principalmente en el yeyuno, seguido del colón, el recto y el estómago. Estos pueden aparecer también fuera del tracto gastrointestinal, incluyendo la pelvis renal, la vejiga, los pulmones y la nasofaringe5. Las características histológicas incluyen: un componente epitelial alargado en forma de onda, una dilatación quística de la glándula que se extiende hacia la submucosa y el músculo liso que se extiende hacia las frondas de los pólipos2.
El diagnóstico clínico se debe considerar en un individuo con al menos uno de los siguientes criterios1-3:
Tres o más pólipos confirmados histológicamente.
Pólipos de Peutz-Jeghers, sin importar la cantidad, en un sujeto con antecedentes familiares del SPJ.
Pigmentación mucocutánea característica en un individuo con antecedentes familiares del SPJ.
Pólipos de Peutz-Jeghers asociados con una pigmentación mucocutánea característica.
Caso clínico
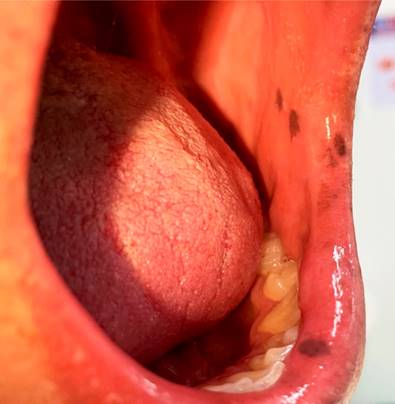
Se presenta el caso de un hombre de 25 años procedente del área urbana del municipio de Barbosa (Santander), quien ingresa al servicio de urgencias por un dolor abdominal difuso de tres años de evolución y sangrado rectal, con antecedente de hemicolectomía derecha por un cuadro de abdomen agudo secundario a intususcepción íleocolónica debido a un pólipo gigante en el íleon terminal de 45 cm x 42 cm. En la pieza quirúrgica también se encontró otro pólipo gigante en el yeyuno, a 80 cm del ángulo de Treitz; ambos con histopatología hamartomatosa. En el examen físico de ingreso se evidenciaron múltiples lesiones maculares hipercromáticas color café oscuro en la mucosa yugal, sin otros hallazgos relevantes (Figura 1).
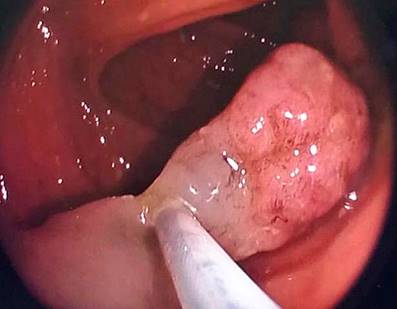
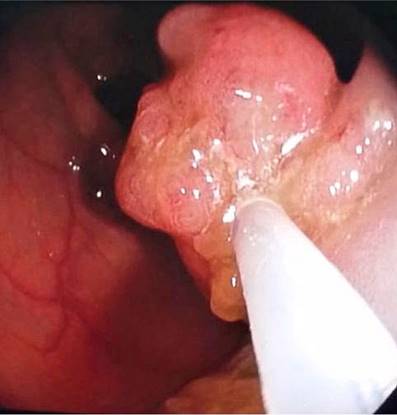
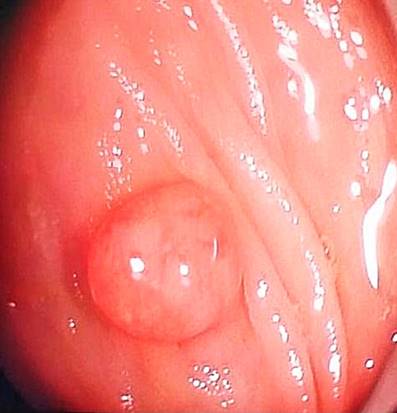
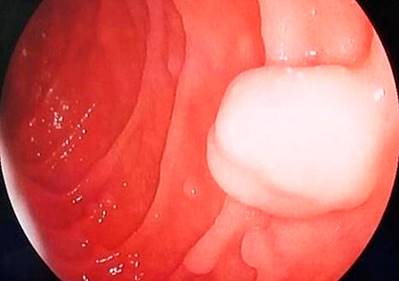
Se hospitalizó al paciente y se le practicó una colonoscopia en la que se observaron dos pólipos a nivel del recto de 7 mm y dos pólipos pediculados en el transverso proximal de 20 mm x 15 mm (Figura 2) y 20 mm x 20 mm (Figura 3), respectivamente. También se le realizó una endoscopia digestiva superior en la que se documentaron múltiples pólipos sésiles (alrededor de 10) entre la primera y la segunda porción duodenal, con diámetros de 4 mm a 12 mm (Figuras 4 y 5). Se realizó una resección endoscópica de todos los pólipos de colon y se resecó uno de los duodenales de mayor tamaño.
El reporte histopatológico de los pólipos de colon fue de tipo hamartomatoso y uno de ellos es un adenoma tubular sin displasia (Figuras 6 y 7). El pólipo de duodeno resecado fue reportado como pólipo hamartomatoso.
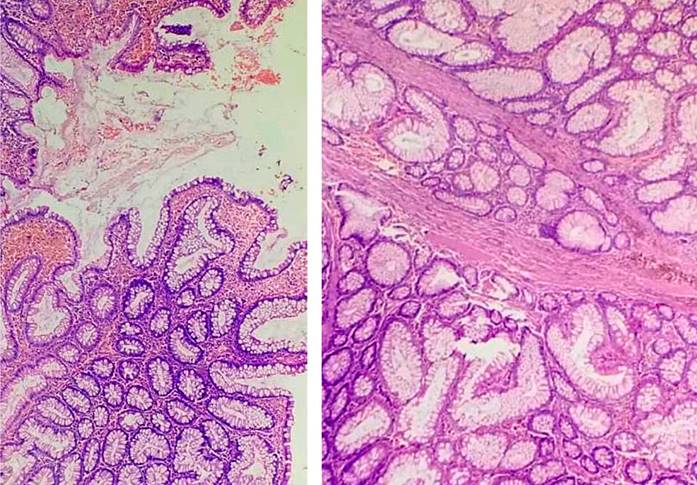
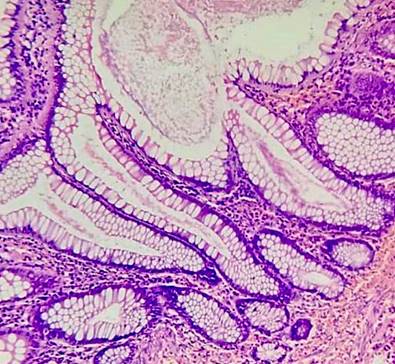
Figura 6 Lesiones polipoideas constituidas por glándulas de diferente forma y tamaño, soportadas por bandas de fibras musculares lisas. Fuente: archivo de los autores.
Discusión
El síndrome de Peutz-Jeghers es una enfermedad autosómica dominante, rara, que tiene pocos reportes en nuestro país. Según Wu y colaboradores, tiene una incidencia estimada de un caso por cada 25.000 a 300.000 nacimientos vivos; puede presentarse en cualquier grupo étnico; afecta a hombres y mujeres por igual; su edad media de diagnóstico es a los 23 años, y la edad promedio para el desarrollo de cáncer es de 42 años6.
Por otra parte, según Beggs y colaboradores, a menudo la primera manifestación es la obstrucción gastrointestinal debida a la invaginación intestinal por los pólipos, un 69% de las veces en el intestino delgado y usualmente ocurre entre los 6 y 18 años2,6.
El objetivo principal en el tratamiento efectivo de los pacientes con el síndrome de Peutz-Jeghers se basa en la vigilancia, la prevención y el tratamiento de las complicaciones. Las recomendaciones incluyen6-8:
○ La detección inicial se realiza a partir de los 12 años.
○ Si se encuentran pólipos, se repite anualmente.
○ En ausencia de pólipos, se repite cada 2-3 años hasta la edad adulta.
○ La detección inicial se realiza a partir de los 12 años o antes si se informan síntomas.
○ Si se encuentran pólipos, se repite anualmente.
○ En ausencia de pólipos, se repite a intervalos de 1-3 años.
○ Colangiopancreatografía por resonancia magnética (CPRM), ecografía endoscópica biliopancreática o ambas: a partir de los 25-30 años; repetir cada 1-2 años.
○ Examen de mamas: examen clínico cada 6 meses a partir de los 25 años.
○ Mamografía: a partir de los 25 años.
Por último, en cuanto a la prevención se recomienda brindar asesoría genética a las parejas que presentan antecedentes familiares del síndrome de Peutz-Jeghers y que desean tener hijos6-10.
Conclusiones
El síndrome de Peutz-Jeghers es una enfermedad poco frecuente, pero con una connotación clínica relevante. Estos pacientes tienen un riesgo 15 veces mayor de malignidad gastrointestinal, aunque también se ha relacionado con neoplasias extraintestinales.
Es de gran importancia conocer su presentación clínica inicial; máculas mucocutáneas hiperpigmentadas (presentes en más del 95% de los casos), antecedentes familiares positivos de síndrome de Peutz-Jeghers y poliposis gastrointestinal de tipo hamartomatoso.
Con lo anterior, se lograría un diagnóstico oportuno para implementar una vigilancia adecuada, de acuerdo con las recomendaciones descritas previamente, con el fin de evitar complicaciones como obstrucción intestinal secundaria a invaginación e intervenciones quirúrgicas de urgencia; y vigilar la aparición de lesiones premalignas y malignas.
REFERENCIAS
1. Daniell J, Plazzer JP, Perera A, Macrae F. An exploration of genotype-phenotype link between Peutz-Jeghers syndrome and STK11: a review. Fam Cancer. 2018;17(3):421-27. https://doi.org/10.1007/s10689-017-0037-3 [ Links ]
2. Beggs A, Latchford A, Vasen H, Moslein G, Alonso A, Aretz S, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975-86. https://doi.org/10.1136/gut.2009.198499 [ Links ]
3. Aguilera Matos I, Díaz Oliva S, Velazco Villaurrutia Yd, García Bacallao E, Labrada Moreno LM. Síndrome de Peutz-Jeghers, experiencia de casos en el Instituto de Gastroenterología. Arch.cuba.gastroenterol. 2019;1(1). Disponible en: http://revgastro.sld.cu/index.php/gast/article/view/18 [ Links ]
4. Parga J, Otero Regino W, Gómez Zuleta M. Prevalencia y características histológicas de los pólipos diminutos del recto y del sigmoides en una población colombiana. Rev. colomb. Gastroenterol. 2020;35(1):25-32. https://doi.org/10.22516/25007440.363 [ Links ]
5. Ospina Nieto J, Pío Quintero Á. Síndrome de Peutz-Jeghers. Presentación de casos y revisión de la literatura. Rev. colomb. Gastroenterol. 2009;24(2):188-99. Disponible en: https://www.redalyc.org/articulo.oa?id=337731592013 [ Links ]
6. Wu M, Krishnamurthy K. Peutz-Jeghers Syndrome. [Actualizado el 21 de agosto de 2022]. En: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK535357/ [ Links ]
7. Signoretti M, Bruno MJ, Zerboni G, Poley JW, Delle Fave G, Capurso G. Results of surveillance in individuals at high-risk of pancreatic cancer: A systematic review and meta-analysis. United European Gastroenterol J. 2018 May;6(4):489-499. https://doi.org/10.1177/2050640617752182 [ Links ]
8. Spoto CPE, Gullo I, Carneiro F, Montgomery EA, Brosens LAA. Hereditary gastrointestinal carcinomas and their precursors: An algorithm for genetic testing. Semin Diagn Pathol. 2018;35(3):170-83. https://doi.org/10.1053/j.semdp.2018.01.004 [ Links ]
9. Grez I, M., Prado A, R., Lahsen H, J. and Hernández M, J. Síndrome de Peutz-Jeghers complicado: Reporte de un caso. Revista chilena de cirugía, vol 60, núm. 3, junio, 2008, pp. 249-254. https://doi.org/10.4067/S0718-40262008000300015 [ Links ]
10. Rodríguez Lagos F, Sorlí Guerola J, Romero Martínez I, Codoñer Franch P. Registro y seguimiento clínico de pacientes con síndrome de Peutz Jeghers en Valencia. Revista de Gastroenterología de México. 2020;85(2):123-139. https://doi.org/10.1016/j.rgmx.2019.02.005 [ Links ]
Citación: Ferreira-Bohórquez EJ, Quintero-Rincón DS, Caro-Gamboa YV, Ayala-Forero MC. Síndrome de Peutz-Jeghers: reporte de caso. Revista. colomb. Gastroenterol. 2022;37(4):502-506. https://doi.org/10.22516/25007440.815
Recibido: 17 de Agosto de 2021; Aprobado: 05 de Noviembre de 2021
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
