











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Cohen (SC) es una enfermedad genética con herencia autosómica recesiva originada por daños en el gen VPS13B (Vacuolar Protein Sorting 13 Homolog B). Se han descrito mutaciones puntuales, deleciones y duplicaciones del locus 8q22-q23 que incluyen este gen y se traducen en alteraciones de una proteína transmembranal de función desconocida1. El fenotipo característico consiste en discapacidad intelectual, microcefalia, facies característica, anormalidades oftálmicas, obesidad central e hipotonía2)(3. Es un síndrome infrecuente, informado por primera vez en 1973 por Cohen y colaboradores quienes describieron el fenotipo original. Se han publicado alrededor de 150 casos, la mayoría en pacientes de origen finlandés, pero también se han reportado casos en Cuba y Brasil4)(5)(6)(7.
El objetivo de esta publicación es presentar un nuevo caso de SC y contribuir a la información epidemiológica sobre este trastorno genético infrecuente, específicamente en Colombia y Latinoamérica, diagnosticado por hibridación genómica comparativa por microarreglos, que mostró una deleción en homocigosis de 0.153 Mb en 8q22.2 incluyendo el gen COH1, OMIM (por la sigla en inglés de Online Mendelian Inheritance in Man) #216550.
REPORTE DEL CASO
Paciente de sexo masculino de 3 años y 10 meses en el momento de la consulta, hijo de padres con relación familiar lejana (Figura 1).
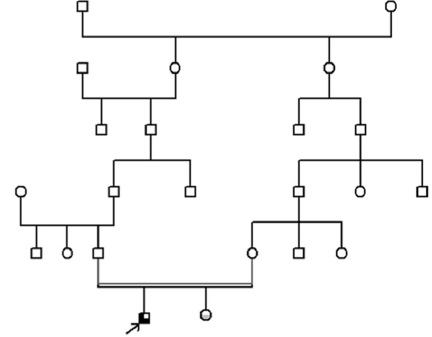
Figura 1 Genealogía. Se observa que el caso aquí reportado es hijo de una unión con parentesco lejano
En el momento del embarazo la madre tenía 42 años, con antecedentes obstétricos de G3 P1 C1; el primer hijo presentó muerte neonatal temprana de causa desconocida; el segundo es una mujer con discapacidad intelectual, facies dismórfica con dientes centrales prominentes y talla baja, hallazgos sugestivos de SC.
En el embarazo del paciente aquí reportado se hizo control prenatal adecuado con cinco ecografías obstétricas que no detectaron anormalidades; por la edad materna se solicitó cariotipo, que no se hizo por decisión de los padres. Nació por cesárea a las 39 semanas por presentación podálica; peso 2600 gramos (-1.65 DS), talla 42 cm (-4.16 DS); observaron deformidad craneal e hicieron tomografía (TAC) que no detectó alteración cerebral; fue dado de alta al día siguiente. A los 3 meses le encontraron hipotonía e iniciaron terapias; a los 8 meses, en una nueva TAC de cráneo, detectaron cierre parcial de las suturas, aumento del diámetro transversal y disminución del anteroposterior, pero sin anomalías cerebrales; se le hizo craneotomía a los 13 meses. En un ecocardiograma a los 3 años no se observaron alteraciones anatómicas. Caminó a los 2,5 años; en el momento de la evaluación solo dice cinco palabras y emite ruidos para comunicarse; no controla esfínteres; los potenciales auditivos son normales, presenta miopía.
Hallazgos relevantes en el examen físico: perímetro cefálico 45 cm (< P3); talla 87 cm (-3.59 DS); peso 12 kg (-2.25 DS); diámetro intercantal externo (DICE) 7,8 cm (+1 DS); diámetro intercantal interno (DICI) 2,8 cm (0 DS); diámetro interpupilar (IP) 4,5 cm (-2 DS). Facies dismórfica caracterizada por depresión bitemporal, fisuras palpebrales estrechas ligeramente inclinadas hacia abajo, pliegue epicántico marcado en el ojo izquierdo, escleróticas grisáceas (Figura 2), incisivos prominentes y micrognatia. Hipotonía marcada, obesidad central insinuada, hipoplasia del pezón derecho, cifoescoliosis, sinostosis radioulnar izquierda, clinodactilia bilateral del quinto dedo (Figura 3). La impresión diagnóstica fue de síndrome dismórfico de probable origen genético. Se hizo hibridación genómica comparativa por microarreglos (HGCm) que mostró una deleción en homocigosis de 0.153 Mb en 8q22.2 que compromete el gen VPS13B y ha sido relacionada con el síndrome de Cohen, OMIM #216550. Se hizo dicho diagnóstico por microdeleción (Figura 4).
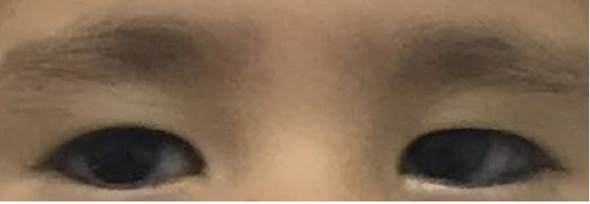
Figura 2 Región periorbitaria. Se observan fisuras palpebrales estrechas ligeramente inclinadas hacia abajo, pliegue epicántico bilateral más marcado en el ojo izquierdo, escleróticas grisáceas y estrabismo.
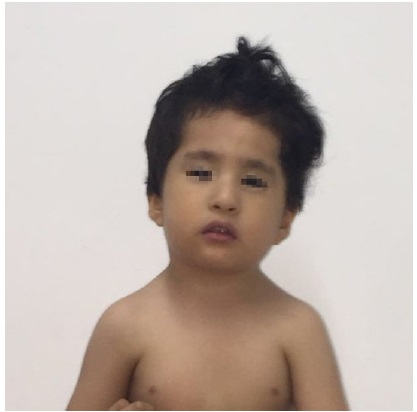
Figura 3 Cara y porción superior del tórax. Se observan signos de hipotonía como la boca abierta y la cabeza inclinada a la izquierda; además, hipoplasia del pezón derecho e incisivos centrales prominentes.
Los dos padres firmaron el consentimiento informado para tomar fotografías y utilizar los datos de la historia clínica.
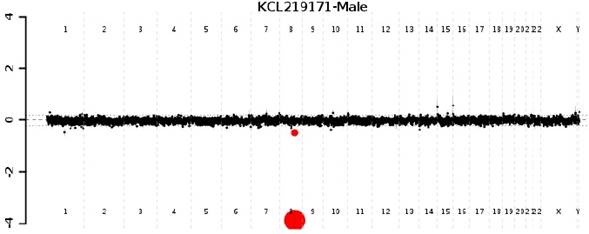
DISCUSIÓN
En 1973, Cohen y colaboradores7 describieron el síndrome que lleva su nombre en dos hermanos y en un tercer paciente, todos con hipotonía, obesidad, incisivos prominentes y retardo mental; posteriormente, Norio y colaboradores8 ampliaron la descripción de los signos a cinco criterios: retardo mental no progresivo acompañado de torpeza motora y microcefalia; rasgos faciales característicos; hipotonía y/o hiperlaxitud articular en la niñez; distrofia retinocoroidea y miopía; neutropenia aislada. Otros rasgos dismórficos que se observan con frecuencia son fisuras palpebrales inclinadas hacia abajo, nariz prominente, surco subnasal (filtrum) corto, boca persistentemente abierta, extremidades delgadas acompañadas de obesidad central en la niñez y comportamiento excesivamente sociable2)(3.
El SC es una enfermedad genética rara; no existen estudios poblacionales de prevalencia; en la literatura solo hay alrededor de 150 casos informados en una amplia variedad de grupos étnicos y se propone que es un síndrome subdiagnosticado4)(9; la mayoría de estos pacientes son de origen finlandés y muestran un fenotipo homogéneo10. Se han publicado series de casos en Bélgica, Líbano, Dinamarca e Inglaterra; en grupos humanos aislados como los Amish hay sobrerrepresentación de la enfermedad con un fenotipo muy similar al informado inicialmente en Finlandia, lo que muestra el carácter recesivo de la enfermedad. Sin embargo, el fenotipo, especialmente los rasgos faciales, varía en otros grupos étnicos, lo que dificulta el diagnóstico basado únicamente en los criterios clínicos11. En Cuba y Brasil se han informado casos de SC con diagnóstico clínico5)(6. El caso aquí publicado sería el primero con diagnóstico molecular en Colombia y Latinoamérica.
El SC es una enfermedad de origen genético dada por daños en el gen VPS13B, antes denominado COH1, en el locus 8q22-2310, que producen cambios en la proteína de su mismo nombre; por homología con modelos en levaduras se le atribuyen funciones relacionadas con el transporte intra- e intercelular de proteínas mediado por vesículas. Estudios funcionales mostraron que la proteína VPS13B se localiza en el aparato de Golgi con la proteína de matriz GM130 y es necesaria para el mantenimiento, el ensamble de las cisternas y la regulación de la formación de túbulos de membrana. La ausencia o alteración de esta proteína puede llevar a la disrupción de la integridad del aparato de Golgi y de su funcionalidad; de igual manera, se han asociado las fallas en esta proteína a defectos de glicosilación y a daños en el mantenimiento de la vía lisosomal-endosomal alterando procesos de degradación intracelular12. No está claro cómo se relacionan los defectos en la proteína VPS13B con los rasgos fenotípicos. Las diferencias en los fenotipos de los pacientes pueden atribuirse a distintas mutaciones en el gen o a las diferencias genéticas entre las poblaciones13.
Mochida y colaboradores14 describieron en 2004 una familia francesa con una mutación que cambiaba el marco de lectura y solo truncaba la región C terminal de la proteína dando lugar a una pérdida parcial en la función de VPS13B por una disrupción de la localización subcelular; este hallazgo se hizo en afectados por SC sin microcefalia. Además, se han descrito mutaciones específicas del gen relacionadas con trastornos del espectro autista y con discapacidad intelectual12.
Dado que en el SC se han documentado diferentes etiologías genéticas como mutaciones puntuales, microdeleciones y microduplicaciones en homocigosis, heterocigosis y en combinación en el gen VPS13B, para el diagnóstico en los pacientes que cumplen con los criterios clínicos de SC2)(3)(8 se han propuesto diferentes técnicas moleculares15.
El Chehadeh y colaboradores15 en 2011 analizaron las publicaciones en que se describen diferentes técnicas moleculares para el diagnóstico de SC como MLPA (por la sigla en inglés de Multiplex ligation-dependent probe amplification), secuenciación del gen e hibridación genómica comparativa por microarreglos de alta resolución (HGCa); concluyeron que al menos en un tercio de las familias en las que se encuentra una causa genética del SC se observan variaciones en el número de copias de oligonucleótidos que incluyen el gen VPS13B y que se pueden detectar mediante HGCa de alta resolución (244K) y que no necesariamente se deben tener sondas específicas. Además, afirmaron que el MLPA con sondas específicas para VPS13B es eficiente para el diagnóstico de SC, pero solamente se hace en muy pocos laboratorios por la baja prevalencia del SC. Propusieron que la primera prueba que se haga en pacientes con sospecha clínica de SC sea la HCGa de alta resolución por la facilidad del acceso a ella en todo el mundo. El diagnóstico del paciente aquí reportado se hizo con dicha prueba y se encontró una deleción de 0.153 Mb que incluía el gen VP13B16.
La HGCm se ha convertido en una herramienta fundamental para el diagnóstico etiológico en individuos con discapacidad intelectual inexplicada, retardo del desarrollo psicomotor, problemas del lenguaje, autismo y anomalías congénitas múltiples; supera las probabilidades de diagnóstico de las pruebas convencionales de citogenética y es fundamental para el enfoque de casos con sospecha de enfermedades de origen genético sin diagnóstico evidente17. Los individuos afectados por el SC cumplen con estas características especialmente antes de los 5 años.
En el diagnóstico diferencial del SC se deben tener en cuenta los síndromes de Barnet-Biedl y Alström que cursan con retardo mental, retinopatía y obesidad; sin embargo, la retinopatía en el SC causa una pérdida de la visión periférica, a diferencia de los síndromes mencionados en los que dicha pérdida es de la visión central. Además, en el síndrome de BarnetBiedl se presentan polidactilia postaxial y displasia renal, y en el de Alström los pacientes generalmente tienen el intelecto normal, sordera, cardiomiopatía y diabetes mellitus; estas características no las tienen los afectados por SC.
El síndrome de glicoproteínas deficientes en carbohidratos tipo 1 es otra enfermedad en la que se presentan retraso del desarrollo, hipotonía, microcefalia, obesidad central y retinopatía pigmentaria; el diagnóstico diferencial con el SC se basa en estudios de imágenes e inmunoensayos en los que la atrofia olivopontocerebelosa y ensayos de actividad de la fosfomanomutasa logran establecer un defecto en el metabolismo de las glicoproteínas2.
No existe un tratamiento específico para los pacientes con SC; dado que tienen bajo peso al nacer, problemas con la alimentación, hipotonía durante la infancia, dificultades para alcanzar los hitos del desarrollo y especialmente retraso en el lenguaje, requieren terapia física, ocupacional y del lenguaje; además, asistir a escuelas especiales donde se pueda atender a sus necesidades educativas. La miopía progresiva y la retinopatía pigmentaria hacen necesarias intervenciones quirúrgicas por el oftalmólogo2.
La consejería genética para los padres de los niños afectados por SC dependerá del daño en el gen que ocasionó el fenotipo; dado que la mayoría de los casos son por herencia autosómica recesiva, la probabilidad de tener un segundo hijo afectado es de 25 %. En términos generales será ideal para los progenitores hacerse pruebas moleculares para ratificar la sospecha de que son portadores de la mutación en heterocigosis. En el caso aquí reportado los padres tenían una relación lejana de consanguinidad y para el momento de la evaluación no tenían expectativa reproductiva, por lo que no se solicitaron pruebas específicas como FISH (por la sigla en inglés de Fluorescence in situ hybridization) de la región 8q22-23.
CONCLUSIONES
Reportamos el caso de un infante con diagnóstico molecular de SC logrado mediante HGCm, en la que se encontró una deleción en homocigosis de 0.153 Mb en el locus 8q22.2 incluyendo el gen COH1, OMIM #216550. Este es el primer caso con diagnóstico molecular reportado en la literatura revisada en Colombia y Latinoamérica. Además, se muestra el aporte de la HGCm para el diagnóstico etiológico en casos de discapacidad intelectual inexplicada, retardo en el desarrollo psicomotor, problemas del lenguaje, autismo y anomalías congénitas múltiples.

