










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957versão On-line ISSN 2500-7440
Rev. colomb. Gastroenterol. vol.37 no.4 Bogotá out./dez. 2022 Epub 07-Jul-2023
https://doi.org/10.22516/25007440.952
Artículo de revisión
Hígado graso (parte 1): aspectos generales, epidemiología, fisiopatología e historia natural

1Specialist in Internal Medicine, Gastroenterology and Hepatology, Universidad Nacional de Colombia, Hospital Clínic de Barcelona, Centro de enfermedades hepáticas y digestivas (CEHYD). Bogotá, Colombia.
2Specialist in Internal Medicine and Gastroenterology, Universidad del Rosario, Universidad Nacional de Colombia. Organización Sanitas, Centro de enfermedades hepáticas y digestivas (CEHYD). Bogotá, Colombia.
3Specialist in Internal Medicine, Gastroenterology and Hepatology, Universidad de Cartagena, Universidad Nacional de Colombia, Hospital Clínic de Barcelona. Head of the Gastroenterology and Hepatology service, Clínica del Norte. Barranquilla, Colombia.
El hígado graso no alcohólico (NAFLD) se define por la presencia de grasa o esteatosis en los hepatocitos y abarca un espectro que va desde la esteatosis simple, pasa por la esteatohepatitis no alcohólica (NASH) con inflamación y fibrosis, y finaliza en la cirrosis. Se considera una prevalencia mundial global cercana al 25% en la población general y se diagnóstica entre los 40 y 50 años, con variaciones respecto al sexo predominante y con diferencias étnicas (la población hispana es la más afectada). El hígado graso está asociado al síndrome metabólico (SM), y la obesidad se considera el principal factor de riesgo con su presencia y con su progresión.
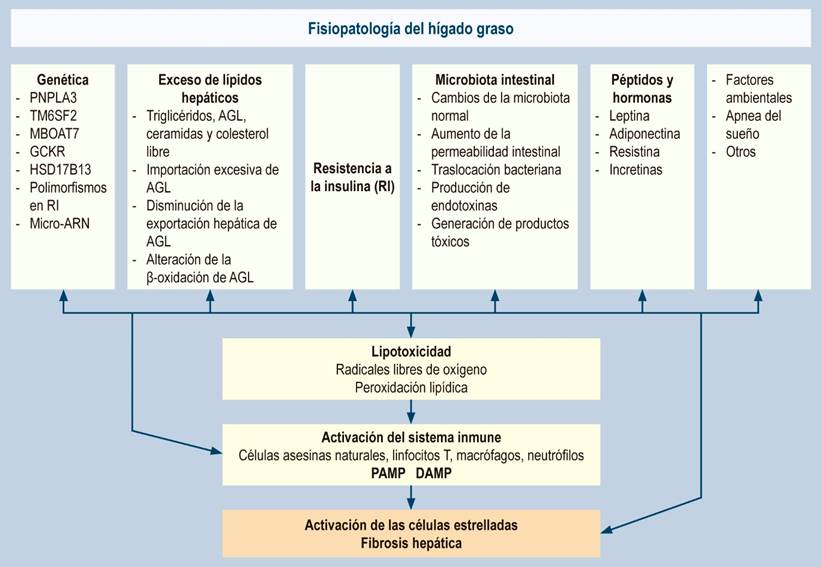
El hígado graso es un trastorno complejo y muy heterogéneo en su fisiopatología, que resulta de la interacción de múltiples elementos: factores genéticos, epigenéticos, ambientales, culturales, entre otros. Todo ello en conjunto lleva a incremento paulatino de grasa hepática, resistencia a la insulina y alteraciones hormonales y de la microbiota intestinal, lo que genera un daño hepatocelular a través de la formación de radicales libres de oxígeno y activación de la fibrogénesis hepática.
La historia natural del hígado graso es dinámica: los pacientes con esteatosis simple tienen bajo riesgo de progresión a cirrosis, mientras que en los pacientes con NASH este riesgo se aumenta; sin embargo, el proceso puede ser reversible y algunas personas tendrán una mejoría espontánea. La fibrosis parece ser el determinante de la mortalidad global y de los desenlaces asociados a la enfermedad hepática; se considera que en todos los pacientes la fibrosis empeora una etapa cada 14 años y en NASH empeora en una etapa cada 7 años. Estudios previos concluyen que aproximadamente 20% de los casos de esteatosis simple progresan a NASH y que, de ellos, aproximadamente el 20% progresan a cirrosis, con presencia de hepatocarcinoma (HCC) en el 5% a 10% de ellos.
Palabras clave: Hígado graso; fisiopatología; historia natural
Fatty liver or NAFLD is defined by the presence of fat or steatosis in hepatocytes and covers a spectrum that goes from simple steatosis, through steatohepatitis (NASH), with inflammation and fibrosis and ending in cirrhosis. It is considered a global world prevalence close to 25% in the general population and is diagnosed between 40 and 50 years, with variations regarding the predominant sex and with ethnic differences, affecting more the Hispanic population. Fatty liver is associated with metabolic syndrome (MS), and obesity is considered the main risk factor for its presence and progression.
Fatty liver is a complex and very heterogeneous disorder in its pathophysiology, resulting from the interaction of multiple elements, genetic, epigenetic, environmental, cultural factors, etc. All this together leads to an accumulation of hepatic fat, insulin resistance, hormonal and intestinal microbiota alterations, generating hepatocellular damage through the formation of free oxygen radicals and activation of hepatic fibrogenesis.
The natural history of fatty liver is dynamic, patients with simple steatosis have a low risk of progression to cirrhosis, in patients with NASH this risk is increased, however, the process may be reversible, and some people will have spontaneous improvement. Fibrosis seems to be the determinant of overall mortality and outcomes associated with liver disease, it is considered that in all patients fibrosis worsens one stage every 14 years, in NASH it worsens one stage every seven years. Previous studies conclude that approximately 20% of cases of simple steatosis progress to NASH and that approximately 20% of them progress to cirrhosis, with the presence of hepatocellular carcinoma (HCC) in 5 to 10% of them.
Keywords: Non-alcoholic Fatty liver disease; pathophysiology; natural history
Introducción
Las primeras observaciones de pacientes con hígado graso datan del siglo XIX1, la enfermedad hepática grasa no alcohólica (NAFLD), o hígado graso, como se le denomina frecuentemente, fue descrita por primera vez por Zelman en 1952, al observar la presencia de enfermedad hepática en pacientes muy obesos2. Ludwig en 1980 describió el término de esteatohepatitis no alcohólica (NASH), al evidenciar una enfermedad con cambios histológicos similares a los observados en pacientes con hepatitis alcohólica, pero con consumo de alcohol insignificante o ausente3, concepto que continua vigente. El hígado graso es la primera causa de consulta en los servicios de hepatología y es frecuente en otras consultas de especialistas clínicos y médicos de atención primaria, debido por un lado, a cambios de hábitos en la vida moderna, dietas hipercalóricas y sedentarismo, y por otro lado, a un mayor acceso a los sistemas de salud, con el uso creciente de ayudas diagnósticas que llevan a frecuentes hallazgos ocasionales de “un hígado graso “en una imagen (usualmente ecografía) o unas transaminasas elevadas en pacientes asintomáticos4, razón por la cual todos los médicos debemos estar preparados para su atención. Por lo anterior se desarrollan estas 2 revisiones de tema.
Definición y diagnóstico
El hígado graso, o NAFLD, se define por la presencia de grasa o esteatosis en los hepatocitos y abarca un espectro que va desde la etapa inicial, la esteatosis simple sin inflamación y fibrosis, pasando por la esteatohepatitis (NASH), con inflamación y fibrosis, hasta la cirrosis, la etapa más avanzada5. Recientemente se ha propuesto el término MAFLD (Metabolic-Associated Fatty Liver Disease) para englobar las alteraciones metabólicas asociadas a hígado graso6. Se debe esperar hasta un consenso con respecto a esta nueva definición que surge, en parte, debido a que el hígado graso es una enfermedad muy heterogénea con múltiples subgrupos.
El hígado graso se diagnostica ante la presencia de esteatosis hepática en cualquier imagen, (usualmente una ecografía), o en una biopsia hepática en el 5% o más del tejido examinado, con o sin inflamación o fibrosis. Se deben descartar causas secundarias de esteatosis hepática como:
Consumo de alcohol mayor de 20 g/día para hombres y mayor de 10 g/día para mujeres.
Ingesta de medicamentos hepatotóxicos en los últimos 6 meses previo al estudio.
Virus de la hepatitis B y C, hemocromatosis, autoinmunidad y otras causas de hepatopatía crónica5.
Epidemiología
Younossi evidenció una prevalencia mundial global cercana al 25% en la población general, con variaciones importantes según la región del mundo evaluada; el Medio Oriente y Suramérica mostraron las mayores prevalencias, 32% y 31%, respectivamente, Norteamérica 24%, Europa 23% y África 13%7. En Estados Unidos, otros estudios han mencionado prevalencias de NAFLD del 10% al 46%8,9. En Latinoamérica, en un estudio mexicano con 2503 individuos se detectó NAFLD en 14,3%, asociado a sobrepeso, obesidad y dislipidemia10. En Chile, otro estudio con 832 pacientes evidenció hígado graso en el 23,4%11. En Colombia no tenemos datos exactos de prevalencia, pero la encuesta nacional de la situación nutricional del 2015 (ENSIN) informó sobrepeso en el 19,1% de los encuestados entre 13 y 17 años y en el 38,4% de los adultos12.
Es importante anotar que la prevalencia de NAFLD ha ido aumentando con el tiempo, hecho demostrado al comparar tres períodos de la Encuesta Nacional de Examen de Salud y Nutrición (NHANES): entre 1988 y 1994, la prevalencia de NAFLD (definida como niveles elevados de aminotransferasas séricas en ausencia de una explicación, lo que podría llevar a subdiagnóstico) fue del 5,5% , entre 1999 y 2004 fue del 9,8% y entre 2005 y 2008 fue del 11%, lo que representa el 47%, 63% y 75% de las enfermedades hepáticas crónicas durante esos períodos. respectivamente13.
El hígado graso se diagnostica usualmente entre los 40 y los 50 años14, con variación respecto al sexo predominante: en unos estudios son las mujeres3,15-17 y en otros, los hombres18-20.
Existen diferencias étnicas en la prevalencia de NAFLD18,21. Un estudio del contenido de triglicéridos hepáticos en 2287 sujetos de una muestra poblacional multiétnica de Estados Unidos encontró una mayor prevalencia de esteatosis hepática en hispanoamericanos (45%) en comparación con blancos (33%) o negros (24%)18. La mayor prevalencia en los hispanos se explicó por una mayor prevalencia de obesidad.
El hígado graso y más la NASH están asociados al síndrome metabólico (SM)5,20-23. La obesidad se considera el principal factor de riesgo, ya que el índice de masa corporal (IMC) y la circunferencia de la cintura se correlacionan positivamente tanto con la presencia de hígado graso como con la progresión de la enfermedad22. El 80% de los pacientes son obesos24,25, 72% presentan dislipidemia25-27, la diabetes tipo 2 (DM2) se encuentra en 45% a 65% de pacientes con NAFLD y se asocia directa y fuertemente con gravedad y progresión del hígado graso5,25-28. La combinación de obesidad, hipertensión sistémica, dislipidemia y resistencia a la insulina o diabetes incrementa el riesgo de fibrosis grave y de enfermedad cardiovascular de forma independiente25,28-30.
El hígado graso es la enfermedad hepática más frecuente en niños y adolescentes obesos y se asocia con trastornos sistémicos crónicos como hipertensión, dislipidemia y mayor riesgo de DM2 y enfermedades cardiovasculares, en la población pediátrica se calcula una prevalencia del hígado graso entre 7,6% y 34%31-33.
Las epidemias de hígado graso y obesidad son un problema de salud pública a nivel mundial5,23,27 y, de hecho, el hígado graso es la indicación de más rápido crecimiento para el trasplante hepático por cirrosis descompensada o HCC34,35.
Fisiopatología
El hígado graso es un trastorno complejo y muy heterogéneo en su fisiopatología, que resulta de la interacción de múltiples elementos, factores genéticos, epigenéticos, ambientales, culturales, entre otros. Todo ello en conjunto produce acumulación de grasa hepática, resistencia a la insulina, alteraciones hormonales y de la microbiota intestinal, lo que genera un daño hepatocelular a través de la formación de radicales libres de oxígeno y activación de la fibrogénesis hepática.
Genética
El primer estudio de asociación del genoma completo en hígado graso (GWAS) mostró la importancia de la herencia en la acumulación de grasa hepática y resaltó la susceptibilidad a la enfermedad según el estado genético de los individuos36; posteriormente, estudios en gemelos confirmaron el componente hereditario de la esteatosis y la fibrosis37, y otros estudios de asociación informaron variantes genéticas asociadas con el riesgo de hígado graso, responsables de codificar proteínas reguladoras del metabolismo de los lípidos hepáticos que llevan a la acumulación de grasa hepática y asociadas al desarrollo y progresión de fibrosis38,39, y cuya expresión fenotípica se desencadena por factores dietéticos y la adiposidad39. Los genes y sus variantes o polimorfismos (SNP) más importantes se presentan a continuación.
Gen PNPLA3
Codifica una fosfolipasa, la adiponutrina, reguladora del metabolismo de los triglicéridos y retinoides33,39. Un polimorfismo de un solo nucleótido (SNP) del gen, el rs738409 (C > G), resulta en una variación de sentido (I148M) que inhibe la enzima mediante un represor que se une competitivamente al coactivador de lipasa adipocítica de triglicéridos (ATGL), lo que provoca una mayor acumulación de lípidos (hasta un 75% más)36,40,41. Los individuos con variante en el nucleótido G tienen un riesgo 3,2 veces mayor de desarrollar fibrosis hepática, y la NASH es más prevalente en los individuos GG que en los CC (odds ratio [OR]: 3,49)41. La variante se ha asociado con esteatohepatitis, fibrosis y cáncer, y se ha encontrado que igualmente promueve el daño hepático en hígado graso alcohólico, hepatitis C crónica e infecciones que promueven la esteatosis hepática39.
La frecuencia de la variante PNPLA3-I148M se correlacionó con el origen étnico y la prevalencia de hígado graso en la población, siendo común en la población mezclada (frecuencia del 26%), más prevalente en población hispana (49%) y menos prevalente en la africana (12-17%)36. Además, la penetrancia de esta variante en la población europea es comparable a los efectos de la mutación de la enfermedad hepática monogénica, con la homocigota GG, que tiene una probabilidad alta hasta 12 veces mayor, de desarrollar HCC en pacientes con NAFLD42,43.
Gen TM6SF2 o miembro 2 de la superfamilia transmembrana 6
El gen regula el contenido de lípidos de los hepatocitos codificando una proteína transmembrana del retículo endoplásmico (RE), asociada con la eliminación de lipoproteínas de muy baja densidad (VLDL)44. Su polimorfismo (SNP), rs58542926 (G > A) que resulta en la variante E167K, se relacionó con niveles altos de triglicéridos hepáticos y riesgo de fibrosis avanzada incrementado45. Por el contrario, esta variante disminuye la secreción de VLDL por los hepatocitos y reduce el riesgo de enfermedad cardiovascular46.
Gen MBOAT7, dominio 7 de O-aciltransferasa unido a la membrana (MBOAT7)
La expresión del gen MBOAT7 produce la enzima lisofosfatidilinositol (LPI) aciltransferasa, una proteína de la endomembrana que cataliza la producción de fosfatidilinositol (PI), componente de las membranas celulares y su SNP rs641738 se ha asociado también con incremento de la fibrosis en hígado graso47,48.
Gen GCKR, gen regulador de la glucocinasa (GCKR)
Se expresa en el hígado y codifica una proteína que actúa como inhibidor alostérico de la GCK, enzima responsable de la homeostasis de la glucosa en sangre. Su variante genética SNP rs780094 se asocia a un mayor nivel sérico de triacilglicerol en ayunas en hígado graso49.
Gen HSD17B13, hidroxiesteroide 17-deshidrogenasa (HSD17B13)
Su SNP rs72613567 es protector y reduce el riesgo en hígado graso, se asoció a niveles reducidos de alanina-aminotransferasa (ALT)50,51.
Base genética de la resistencia a la insulina
Se ha asociado con polimorfismos en el gen de la apolipoproteína C3, interleucina 6 (IL-6) y adiponutrina52-55, con alteraciones en la actividad transcripcional del promotor del coactivador 1-alfa del receptor gamma activado por el proliferador de peroxisomas (PPARGC1A)52; adicionalmente, otras citocinas y adipocinas involucradas en la señalización del receptor de insulina parecen estar alteradas en el tejido adiposo omental de los pacientes con NASH55.
Otros elementos genéticos: ARN no codificantes (ARNnc)
Son ARN que no codifican proteínas funcionales y se agrupan según su tamaño, pequeños (micro-ARN) y grandes, incluidos los ARN largos y los circulares. Regulan múltiples vías biológicas, captación de lípidos, lipogénesis de novo, oxidación de lípidos, exportación de lípidos hepáticos, apoptosis, proliferación celular o fibrosis56. Por lo menos una docena de micro-ARN se han asociado fuertemente con el desarrollo de hígado graso56,57. A medida que aumenta la muerte celular con la progresión de la esteatohepatitis, se liberan directamente o empaquetados en exosomas a la circulación, lo que también los convierte en biomarcadores potenciales para el estado del hígado graso58.
Acúmulo excesivo de lípidos en el hígado
El desequilibrio hepático en la entrada y salida de grasas, debido a cambios en el estilo de vida por el alto consumo de calorías, sedentarismo, síndrome metabólico, alteraciones hormonales y genéticas resulta en la esteatosis hepática con exceso de triglicéridos, ácidos grasos libres (AGL), ceramidas y colesterol libre. Esto puede ocurrir por la importación excesiva de AGL del tejido adiposo, por una disminución de la exportación hepática de AGL (secundaria a una reducida síntesis o secreción de VLDL), o por una alteración de la β-oxidación de AGL. Las principales fuentes de triglicéridos son los ácidos grasos almacenados en el tejido adiposo y los ácidos grasos recién elaborados en el hígado mediante lipogénesis de novo59,60. Adicionalmente, se ha informado que la lipogénesis de novo (LDN) está regulada al alza en los pacientes con hígado graso, es tres veces mayor en comparación con los pacientes de control y esta vía puede ser patogénicamente más relevante en pacientes mayores y hombres61,62.
El aumento del tejido adiposo visceral y la grasa intrahepática se correlacionan con un aumento de la gluconeogénesis, un aumento de los niveles de AGL y resistencia a la insulina; la grasa visceral también se ha asociado con inflamación y fibrosis hepática en pacientes con NASH independientemente de la resistencia a la insulina, un efecto posiblemente mediado por la IL-6 (citocina proinflamatoria)63,64.
Resistencia a la insulina
Es uno de los mecanismos básicos en la fisiopatología del hígado graso, se presenta en la mayoría de los pacientes obesos y diabéticos, aunque se puede encontrar también en pacientes delgados no diabéticos65-67. La resistencia a la insulina desencadena múltiples eventos en el metabolismo de los lípidos: aumento en la lipólisis periférica y en la síntesis de triglicéridos, mayor captación hepática de AGL y, por todo lo anterior, acumulación de triglicéridos en los hepatocitos, lo que a su vez resulta en un cambio preferencial de los carbohidratos a la β-oxidación de AGL65,68,69. Las vías moleculares que conducen a la resistencia a la insulina son complejas y no se han dilucidado por completo, pero varias moléculas parecen interferir con las acciones de la insulina a nivel celular; por ejemplo, se ha demostrado que los ácidos biliares lipofílicos promueven la sensibilidad a la insulina y disminuyen la gluconeogénesis y la trigliceridemia hepática mediante la unión al receptor nuclear farnesoide X70.
Microbiota intestinal
El microbioma intestinal humano se compone de 10 a 100 billones de microorganismos, principalmente bacterias; tiene diez veces más microorganismos intestinales que células eucariotas humanas71 y es susceptible a alteraciones ambientales y fisiopatológicas, por lo cual desempeña un papel fundamental en la fisiopatología del hígado graso. La microbiota se ha asociado a lesión directa e indirecta de la célula hepática a través de varios mecanismos que producen lipotoxicidad, daño oxidativo y fibrosis secundaria72-79:
Cambios de la microbiota normal: sobrecrecimiento bacteriano en el intestino delgado o cambios en la composición de la microbiota intestinal (por ejemplo, composición de macronutrientes en dietas ricas en fructosa y grasas saturadas).
Aumento de la permeabilidad intestinal asociada posiblemente a sobrecrecimiento bacteriano del intestino delgado, cambios en la composición de la microbiota y traslocación bacteriana.
Mayor producción de endotoxinas.
Generación de productos tóxicos como el alcohol y acetaldehído endógenos a nivel colónico por bacterias y levaduras, y otros metabolitos de la microbiota intestinal como el ácido N, N, N-trimetil-5-aminovalérico (TMAVA), que reduce la síntesis de carnitina y la oxidación de ácidos grasos hepáticos, y lleva a esteatosis hepática.
Deconjugación de sales biliares e inactivación de los lipótropos hepáticos, como la colina.
En la disbiosis intestinal, definida como el desequilibrio entre las comunidades microbianas residentes y el huésped, un aumento de la permeabilidad intestinal puede conducir a una translocación patológica de productos microbianos al hígado a través de la vena porta73-75; en este proceso se producen patrones moleculares asociados a patógenos (PAMP), que son reconocidos por receptores selectivos en el hígado, principalmente receptores tipo Toll, y generan una respuesta inmunitaria innata crónica que libera patrones moleculares asociados al daño (DAMP)80. Además, los metabolitos derivados de la microbiota, como los ácidos biliares modificados, la colina y el etanol, alteran el metabolismo hepático y desencadenan una respuesta inflamatoria72,75,77-80.
Péptidos y hormonas
Leptina
Es un péptido producido en el tejido adiposo, su ausencia se asocia con obesidad masiva en ratones (ob/ob) y en humanos. La leptina induce la desfosforilación del sustrato 1 del receptor de insulina, lo que hace que los hepatocitos sean más resistentes a la insulina81. Parece que la resistencia a la leptina en el sistema nervioso central, más que en el hígado, puede ser importante en la patogenia de la NASH, lo que se dedujo al observar que la infusión de leptina en el sistema nervioso central de ratones con hígado graso corrigió la resistencia a la insulina y el hígado graso, mientras que la administración periférica no lo hizo82.
Adiponectina
Es otra hormona secretada únicamente en el tejido adiposo con efectos beneficiosos en el metabolismo de los lípidos, mejora la β-oxidación de los ácidos grasos en el músculo y la depuración de lípidos plasmáticos, además mediante la inhibición de la producción del factor de necrosis tumoral alfa (TNF-α) en el hígado, produce efectos antiinflamatorios directos83. La adiponectina parece tener alguna relación con la modulación de la sensibilidad a la insulina y niveles circulantes bajos de la hormona se han correlacionado con la gravedad de los hallazgos histopatológicos en NASH84.
Resistina
Es una proteína derivada del tejido adiposo asociada al desarrollo de resistencia a la insulina. En una investigación con ratones, la sobreexpresión de resistina llevó a niveles de AGL alterados, intolerancia a la glucosa e hiper-insulinemia85.
Incretinas
Son hormonas derivadas del intestino, como el polipéptido insulinotrópico dependiente de glucosa (GIP) y el péptido similar al glucagón 1 (GLP-1), que potencian la secreción de insulina después de la ingestión de alimentos y desempeñan un papel importante en la homeostasis de la glucosa86.
Otros factores asociados propuestos
Factores ambientales
Los factores de riesgo modificables, como el trabajo por turnos y los viajes que perturban la alimentación normal y los ciclos de sueño y vigilia, promueven la adiposidad, el síndrome metabólico y la enfermedad del hígado graso no alcohólico87. La interrupción prolongada de los ritmos circadianos normales en ratones con hígado graso induce el desarrollo de esteatohepatitis no alcohólica al desregular la comunicación cruzada entre dos receptores de hormonas nucleares: el receptor X farnesoide (FXR) y el receptor de androstano constitutivo (CAR), lo que lleva a la supresión de FXR, acumulación hepática de ácidos biliares, sobreactivación de CAR inducida por ácidos biliares y eventual lesión hepática, fibrosis y neoplasia dependientes del CAR88-90. También se han implicado como factores patogénicos las toxinas ambientales91 y la apnea obstructiva del sueño, que generan inflamación92,93.
Daño hepatocelular
Se produce finalmente por la formación de radicales libres generados por:
Inducción de lipoxigenasas microsomales del citocromo p-450, a partir de los AGL.
El cambio a β-oxidación de los AGL más defectos preexistentes en la fosforilación oxidativa mitocondrial (anomalías estructurales mitocondriales significativas), que se han demostrado por microscopía electrónica de hepatocitos de pacientes con NASH, no observados en pacientes con esteatosis hepática simple22,64-70.
Estas 2 vías conjuntamente llevan a daño hepatocelular y fibrosis, mediante la activación de múltiples procesos como el factor nuclear kappa B (NF-κB), aumento en la producción de citocinas, activación del TNF-α, el sistema del complemento, la mieloperoxidasa plasmática y las células asesinas naturales65,94-96.
La formación de radicales libres de oxígeno y la peroxidación lipídica pueden agotar las enzimas antioxidantes como el glutatión, la vitamina E, el betacaroteno y la vitamina C, lo que vuelve al hepatocito más susceptible a la lesión oxidativa, y en este proceso los niveles séricos de xantina oxidasa que producen las especies reactivas de oxígeno se encuentran aumentados al compararlos con pacientes controles, mientras que, por el contrario, los niveles de múltiples enzimas antioxidantes se encuentran disminuidos97,98.
Se ha descrito una correlación entre la gravedad de la enfermedad y el aumento de la expresión de los receptores depuradores oxidativos99. La serotonina se ha implicado como fuente de especies reactivas de oxígeno en NASH, el aumento del catabolismo de la serotonina, resultó en un aumento de los niveles de especies oxidativas reactivas y necroinflamación en un modelo animal100.
El hierro contribuye al daño hepatocelular mediante la formación de especies de radicales libres de oxígeno generados en su proceso de reducción de Fe3+ a Fe2+(101, asociado al desarrollo de NASH; también se ha observado un aumento del hierro en los pacientes con resistencia a la insulina de forma independiente102. Una mayor concentración de hierro hepático parece correlacionarse con la gravedad de la fibrosis en la NASH103.
El sistema inmunitario innato juega un papel en la progresión del hígado graso104,105. Una respuesta inmunitaria fisiológica y adecuada es fundamental para la resolución de un daño hepático y la regeneración normal del hígado, mientras que una respuesta exagerada y persistente del sistema inmunitario innato puede conducir a una inflamación crónica del hígado y a las consecuencias de ello. Los PAMP y los DAMP activan y mantienen una respuesta inmunitaria innata mediada por un receptor de reconocimiento de patrones, característica del hígado graso y una compleja comunicación cruzada intercelular entre las diferentes células inmunitarias innatas, células asesinas naturales, linfocitos T, macrófagos, neutrófilos, hepatocitos y células estrelladas hepáticas (HSC); el resultado de esto define la progresión a esteatohepatitis y fibrosis105.
Fuera de la muerte de los hepatocitos por la lipotoxicidad directa y la liberación de DAMP, se ha identificado la piroptosis como una nueva forma de muerte celular programada en hígado graso, caracterizada por la activación y ensamblaje de complejos multiproteicos, llamados inflamasomas106,107, el más estudiado es el dominio de unión a nucleótidos, una familia de repeticiones ricas en leucina (NLR), dominio de pirina que contiene 3 inflamasomas (NLRP3). Una vez activado, el inflamasoma NLRP3 escinde la procaspasa 1 a su forma madura (caspasa-1) que posteriormente escinde IL-1β, IL-18 y gasdermina D. La IL-1 y la IL-18 son citocinas altamente proinflamatorias que se liberan en el espacio extracelular en parte a través de los poros transmembrana formados por gasdermina D108,109. Todos los factores fisiopatológicos se resumen en la Figura 1.
La fibrosis hepática es la característica patológica que predice los diferentes resultados asociados a la lesión hepática en el hígado graso110,111 y su génesis, fibrogénesis hepática o formación de cicatriz; es un proceso dinámico de acumulación de matriz extracelular (MEC) en la que la célula estrellada hepática (anteriormente denominada lipocito, célula de Ito, o célula perisinusoidal) es su principal fuente en el hígado normal y fibrótico112.
Un daño hepático crónico estimula de forma paracrina las células vecinas, como las células endoteliales sinusoidales, células de Kupffer, monocitos y plaquetas, entre otras, lo que produce un estímulo angiogénico con formación de nuevos vasos sanguíneos, remodelación sinusoidal y liberación de múltiples mediadores como el factor de crecimiento derivado de plaquetas (PDGF), el factor de crecimiento del endotelio vascular (VEGF) y sus receptores afines, mediadores vasoactivos que incluyen el óxido nítrico y el monóxido de carbono113. Todo ello en conjunto produce una activación de las células estrelladas que aumentan la producción de colágenos (tipos I, III y IV), varias glicoproteínas (fibronectina celular, laminina, osteonectina, tenascina, factor de Von Willebrand), proteoglicanos y glucosaminoglicanos (perlecán, decorina, entre otras), llevando a una acumulación progresiva y un cambio en la composición de la MEC, que activa vías de retroalimentación positiva y amplifican aún más la fibrosis mediante procesos que involucran a los receptores de la membrana celular y a una amplia variedad de otras citocinas y proteínas de adhesión. Una de estas vías bien caracterizada es la de las integrinas, una familia de proteínas de membrana que controlan varias funciones celulares, incluida la expresión génica, el crecimiento y la diferenciación celular114.
Por su parte, la fibrosis refleja un equilibrio entre la producción y la degradación de la MEC, un delicado balance entre enzimas dependientes de calcio que degradan específicamente colágenos y sustratos no colágenos, conocidas como metaloproteinasas de la matriz (MMP, o matrixinas) y sus respectivos inhibidores específicos, conocidos como inhibidores tisulares de metaloproteinasas (TIMP). La sustitución de la matriz de baja densidad por la de tipo intersticial, característica de la fibrosis, tiene consecuencias sobre la función de los hepatocitos, las células estrelladas hepáticas y las células endoteliales, lo que explica en parte la disfunción sintética y metabólica observada en pacientes con fibrosis avanzada112-114.
Historia natural
Estudios de biopsia pareados evidenciaron que la historia natural del hígado graso es muy dinámica: los pacientes con esteatosis simple tienen bajo riesgo de progresión a cirrosis, mientras que en los pacientes con NASH este riesgo aumenta; sin embargo, el proceso puede ser reversible y algunas personas tendrán una mejoría espontánea16,115-120. La fibrosis parece ser el determinante de la mortalidad global y de los desenlaces asociados a la enfermedad hepática113,114,121,122. El daño causado por NAFLD puede tardar muchos años en progresar y se describe con mayor frecuencia en 5 etapas, de 0 a 4, según la cantidad y el patrón de distribución de la fibrosis encontrada, semejante a la clasificación METAVIR (F0-F4)123.
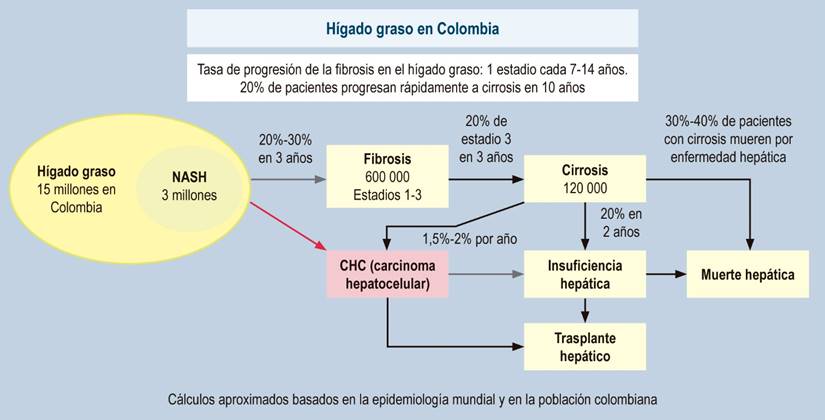
En hígado graso, teniendo en cuenta que los datos son cifras promedio y que no son lineales, se considera que en todos los pacientes la fibrosis empeora una etapa cada 14 años, mientras que en NASH empeora una etapa cada 7 años120. Estudios previos concluyen que aproximadamente 20% de los casos de esteatosis simple progresan a NASH y que, de ellos, aproximadamente el 20% progresan a cirrosis, con presencia de hepatocarcinoma (HCC) en el 5% a 10% de ellos120,124-126. Análisis recientes mostraron que el 20% de los pacientes con F3 progresaron a cirrosis en 2 años y, de ellos, el 20% de los pacientes con cirrosis compensada se descompensaron127,128. Estas cifras se denominan “la regla” del 20% y corroboran los datos anteriores. En algunos casos, el hígado puede dañarse mucho más rápido que estas cifras promedio y hasta 1 de cada 5 pacientes con fibrosis son progresores rápidos120; esto podría deberse a fluctuaciones en la gravedad de los factores de riesgo metabólicos, así como al impacto de diversos estilos de vida (poco saludables), además de factores genéticos. En la actualidad, se reconoce que la NASH es la principal causa de cirrosis criptogénica (Figura 2)129. Teniendo en cuenta que en Colombia no hay datos exactos de nuestra historia natural y con base en los datos internacionales, en la Figura 3 planteamos una proyección de la enfermedad para nuestro país.
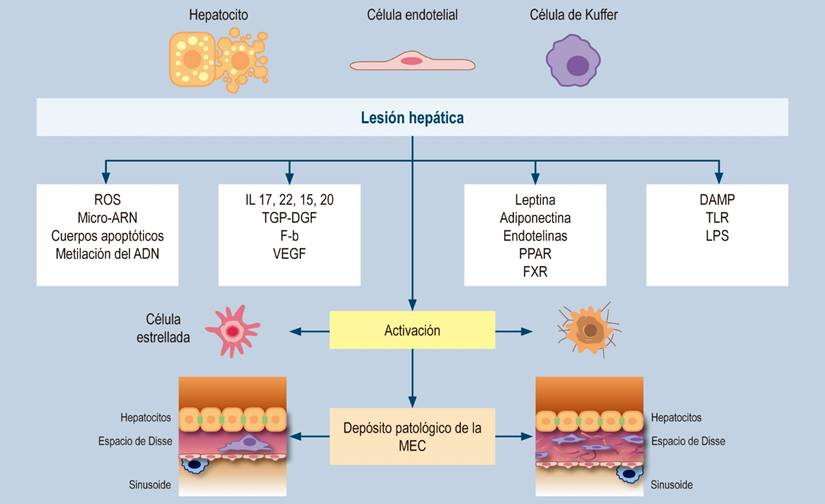
Figura 2 Fibrogénesis hepática. Modificado de: Tsuchida T et al. 2017;14(7):397-411; Friedman SL. Nat Rev Gastroenterol Hepatol. 2010;7(8):425-36; Kisseleva T et al. Nat Rev Gastroenterol Hepatol. 2021;18(3):151-166.
REFERENCIAS
1. Ayonrinde OT. Historical narrative from fatty liver in the nineteenth century to contemporary NAFLD - Reconciling the present with the past. JHEP Rep. 2021;3(3):100261. https://doi.org/10.1016/j.jhepr.2021.100261 [ Links ]
2. Zelman S. The liver in obesity. AMA Arch Intern Med. 1952;90(2):141-56. https://doi.org/10.1001/archinte.1952.00240080007002 [ Links ]
3. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55(7):434-8. [ Links ]
4. Patel V, Sanyal AJ, Sterling R. Clinical Presentation and Patient Evaluation in Nonalcoholic Fatty Liver Disease. Review Clin Liver Dis. 2016;20(2):277-92. https://doi.org/10.1016/j.cld.2015.10.006 [ Links ]
5. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328-357.https://doi.org/10.1002/hep.29367 [ Links ]
6. Eslam M, Sanyal AJ, George J; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology. 2020;158(7):1999-2014.e1.https://doi.org/10.1053/j.gastro.2019.11.312 [ Links ]
7. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11-20. https://doi.org/10.1038/nrgastro.2017.109 [ Links ]
8. Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124-31. https://doi.org/10.1053/j.gastro.2010.09.038 [ Links ]
9. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34(3):274-85. https://doi.org/10.1111/j.1365-2036.2011.04724.x [ Links ]
10. Lizardi-Cervera J, Laparra IB, Chávez-Tapia NC, Ostos MER, Uribe Esquivel M. Prevalencia de hígado graso no alcohólico y síndrome metabólico en población asintomática. Rev Gastroenterol México 2006;71(4):453-9. [ Links ]
11. Riquelme A, Arrese M, Soza A, Morales A, Baudrand R, Pérez-Ayuso RM, et al. Non-alcoholic fatty liver disease and its association with obesity, insulin resis-tance and increased serum levels of C-reactive protein in Hispanics. Liver Int. 2009;29(1):82-8. https://doi.org/10.1111/j.1478-3231.2008.01823.x [ Links ]
12. Encuesta Nacional de la Situación Nutricional ENSIN 2015. Colombia: Ministerio de Salud y Protección Social; 2015. [ Links ]
13. Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9(6):524-530.e1; quiz e60. https://doi.org/10.1016/j.cgh.2011.03.020 [ Links ]
14. Falck-Ytter Y, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis. 2001;21(1):17-26. https://doi.org/10.1055/s-2001-12926 [ Links ]
15. Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999;30(6):1356-62. https://doi.org/10.1002/hep.510300604 [ Links ]
16. Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116(6):1413-9. https://doi.org/ 0.1016/s0016-5085(99)70506-8 [ Links ]
17. Cortez-Pinto H, Camilo ME, Baptista A, De Oliveira AG, De Moura MC. Non-alcoholic fatty liver: another feature of the metabolic syndrome? Clin Nutr. 1999;18(6):353-8. https://doi.org/10.1016/S0261-5614(99)80015-6 [ Links ]
18. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387-95. https://doi.org/10.1002/hep.20466 [ Links ]
19. Arun J, Clements RH, Lazenby AJ, Leeth RR, Abrams GA. The prevalence of nonalcoholic steatohepatitis is greater in morbidly obese men compared to women. Obes Surg. 2006;16(10):1351-8. https://doi.org/10.1381/096089206778663715 [ Links ]
20. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37(4):917-23. https://doi.org/10.1053/jhep.2003.50161 [ Links ]
21. Weston SR, Leyden W, Murphy R, Bass NM, Bell BP, Manos MM, et al. Racial and ethnic distribution of nonalcoholic fatty liver in persons with newly diagnosed chronic liver disease. Hepatology. 2005;41(2):372-9. https://doi.org/10.1002/hep.20554 [ Links ]
22. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346(16):1221-31. https://doi.org/10.1056/NEJMra011775 [ Links ]
23. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263-73. https://doi.org/10.1001/jama.2015.5370 [ Links ]
24. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol. 2019;71(04):793-801. https://doi.org/10.1016/j.jhep.2019.06.021 [ Links ]
25. Rinella M, Charlton M. The globalization of nonalcoholic fatty liver disease: prevalence and impact on world health. Hepatology. 2016;64(1):19-22. https://doi.org/10.1002/hep.28524 [ Links ]
26. Jarvis H, Craig D, Barker R, Spiers G, Stow D, Anstee QM, et al. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of population-based observational studies. PLoS Med. 2020;17(4):e1003100. https://doi.org/10.1371/journal.pmed.1003100 [ Links ]
27. Arab JP, Dirchwolf M, Álvares-da-Silva MR, Barrera F, Benítez C, Castellanos-Fernandez M, et al. Latin American Association for the study of the liver (ALEH) practice guidance for the diagnosis and treatment of non-alcoholic fatty liver disease. Ann Hepatol. 2020;19(6):674-690. https://doi.org/10.1016/j.aohep.2020.09.006 [ Links ]
28. Loomba R, Abraham M, Unalp A, Wilson L, Lavine J, Doo E, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56(3):943-51.https://doi.org/10.1002/hep.25772 [ Links ]
29. Ma J, Hwang SJ, Pedley A, Massaro JM, Hoffmann U, Chung RT, et al. Bi-directional analysis between fatty liver and cardiovascular disease risk factors. J Hepatol. 2017;66(2):390-397.https://doi.org/10.1016/j.jhep.2016.09.022 [ Links ]
30. Stepanova M, Younossi ZM. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin Gastroenterol Hepatol. 2012;10(6):646-50. https://doi.org/10.1016/j.cgh.2011.12.039 [ Links ]
31. Giorgio V, Prono F, Graziano F, Nobili V. Pediatricnon alcoholic fatty liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. 2013;13:40. https://doi.org/10.1186/1471-2431-13-40 [ Links ]
32. Anderson EL, Howe LD, Jones HE, Higgins JPT, Lawlor DA, Fraser A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS One. 2015;10(10):e0140908. https://doi.org/10.1371/journal.pone.0140908 [ Links ]
33. Yu EL, Golshan S, Harlow KE, Angeles JE, Durelle J, Goyal NP, et al. Prevalence of Nonalcoholic Fatty Liver Disease in Children with Obesity. J Pediatr. 2019;207:64-70. https://doi.org/10.1016/j.jpeds.2018.11.021 [ Links ]
34. Majumdar A, Tsochatzis EA. Changing trends of liver transplantation and mortality from non-alcoholic fatty liver disease. Metabolism. 2020;111S:154291.https://doi.org/10.1016/j.metabol.2020.154291 [ Links ]
35. Younossi ZM, Marchesini G, Pinto-Cortez H, Petta S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation. 2019;103(1):22-27. https://doi.org/10.1097/TP.0000000000002484 [ Links ]
36. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461-5. https://doi.org/10.1038/ng.257 [ Links ]
37. Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology. 2015;149(7):1784-93. https://doi.org/10.1053/j.gastro.2015.08.011 [ Links ]
38. Jonas W, Schürmann A. Genetic and epigenetic factors determining NAFLD risk. Mol Metab. 2021;50:101111. https://doi.org/10.1016/j.molmet.2020.101111 [ Links ]
39. Carlsson B, Lindén D, Brolén G, Liljeblad M, Bjursell M, Romeo S, et al. Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2020;51(12):1305-1320. https://doi.org/10.1111/apt.15738 [ Links ]
40. Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology. 2019;69(6):2427-2441. https://doi.org/10.1002/hep.30583 [ Links ]
41. Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883-94. https://doi.org/10.1002/hep.24283 [ Links ]
42. Krawczyk M, Stokes CS, Romeo S, Lammert F. HCC and liver disease risks in homozygous PNPLA3 p.I148M carriers approach monogenic inheritance. J Hepatol. 2015;62(4):980-1. https://doi.org/10.1016/j.jhep.2014.10.048 [ Links ]
43. Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2014;61(1):75-81. https://doi.org/10.1016/j.jhep.2014.02.030 [ Links ]
44. Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet. 2017;26(14):2719-2731. https://doi.org/10.1093/hmg/ddx159 [ Links ]
45. Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309. https://doi.org/10.1038/ncomms5309 [ Links ]
46. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61(2):506-14.https://doi.org/10.1002/hep.27490 [ Links ]
47. Caddeo A, Jamialahmadi O, Solinas G, Pujia A, Mancina RM, Pingitore P, et al. MBOAT7 is anchored to endomembranes by six transmembrane domains. J Struct Biol. 2019;206(3):349-360. https://doi.org/10.1016/j.jsb.2019.04.006 [ Links ]
48. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology. 2016;150(5):1219-1230.e6. https://doi.org/10.1053/j.gastro.2016.01.032 [ Links ]
49. Zain SM, Mohamed Z, Mohamed R. Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. 2015;30(1):21-7. https://doi.org/10.1111/jgh.12714 [ Links ]
50. Luukkonen PK, Tukiainen T, Juuti A, Sammalkorpi H, Haridas PAN, Niemelä O, et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight. 2020;5(5):e132158. https://doi.org/10.1172/jci.insight.132158 [ Links ]
51. Yang J, Trépo E, Nahon P, Cao Q, Moreno C, Letouzé E, et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology. 2019;70(1):231-240. https://doi.org/10.1002/hep.30623 [ Links ]
52. Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology. 2010;52(6):1992-2000. https://doi.org/10.1002/hep.23927 [ Links ]
53. Dongiovanni P, Valenti L, Rametta R, Daly AK, Nobili V, Mozzi E, et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut. 2010;59(2):267-73. https://doi.org/10.1136/gut.2009.190801 [ Links ]
54. Petersen KF, Dufour S, Hariri A, Nelson-Williams C, Foo JN, Zhang XM, et al. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med. 2010;362(12):1082-9. https://doi.org/10.1056/NEJMoa0907295 [ Links ]
55. Carulli L, Canedi I, Rondinella S, Lombardini S, Ganazzi D, Fargion S, et al. Genetic polymorphisms in non-alcoholic fatty liver disease: interleukin-6-174G/C polymorphism is associated with non-alcoholic steatohepatitis. Dig Liver Dis. 2009;41(11):823-8. https://doi.org/10.1016/j.dld.2009.03.005 [ Links ]
56. Shabgah AG, Norouzi F, Hedayati-Moghadam M, Soleimani D, Pahlavani N, Navashenaq JG. A comprehensive review of long non-coding RNAs in the pathogenesis and development of non-alcoholic fatty liver disease. Nutr Metab (Lond). 2021;18(1):22. https://doi.org/10.1186/s12986-021-00552-5 [ Links ]
57. Tsagakis I, Douka K, Birds I, Aspden JL. Long non-coding RNAs in development and disease: conservation to mechanisms. J Pathol. 2020;250(5):480-495. https://doi.org/10.1002/path.5405 [ Links ]
58. Khalifa O, Errafii K, Al-Akl NS, Arredouani A. Noncoding RNAs in Nonalcoholic Fatty Liver Disease: Potential Diagnosis and Prognosis Biomarkers. Dis Markers. 2020;2020:8822859. https://doi.org/10.1155/2020/8822859 [ Links ]
59. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68(2):280-295. https://doi.org/10.1016/j.jhep.2017.11.014 [ Links ]
60. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343-51. https://doi.org/10.1172/JCI23621 [ Links ]
61. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726-35. https://doi.org/10.1053/j.gastro.2013.11.049 [ Links ]
62. Pramfalk C, Pavlides M, Banerjee R, McNeil CA, Neubauer S, Karpe F, et al. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J Clin Endocrinol Metab. 2015;100(12):4425-33. https://doi.org/10.1210/jc.2015-2649 [ Links ]
63. Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133(2):496-506. https://doi.org/10.1053/j.gastro.2007.04.068 [ Links ]
64. van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, et al. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology. 2008;48(2):449-57. https://doi.org/10.1002/hep.22350 [ Links ]
65. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183-92. https://doi.org/10.1053/gast.2001.23256 [ Links ]
66. Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35(2):373-9. https://doi.org/10.1053/jhep.2002.30692 [ Links ]
67. Willner IR, Waters B, Patil SR, Reuben A, Morelli J, Riely CA. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol. 2001;96(10):2957-61. https://doi.org/10.1111/j.1572-0241.2001.04667.x [ Links ]
68. Kral JG, Lundholm K, Björntorp P, Sjöström L, Scherstén T. Hepatic lipid metabolism in severe human obesity. Metabolism. 1977;26(9):1025-31. https://doi.org/10.1016/0026-0495(77)90020-8 [ Links ]
69. Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest. 1983;72(5):1737-47. https://doi.org/10.1172/JCI111133 [ Links ]
70. Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53(9):1723-37. https://doi.org/10.1194/jlr.R024794 [ Links ]
71. Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab. 2013;24(11):537-45. https://doi.org/10.1016/j.tem.2013.05.009 [ Links ]
72. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1877-87. https://doi.org/10.1002/hep.22848 [ Links ]
73. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63(3):764-75. https://doi.org/10.1002/hep.28356 [ Links ]
74. Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology. 2018;68(3):918-932. https://doi.org/10.1002/hep.29892 [ Links ]
75. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017;25(5):1054-1062.e5. https://doi.org/10.1016/j.cmet.2017.04.001 [ Links ]
76. Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology. 2016;151(4):733-746.e12. https://doi.org/10.1053/j.gastro.2016.06.022 [ Links ]
77. Cope K, Risby T, Diehl AM. Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology. 2000;119(5):1340-7. https://doi.org/10.1053/gast.2000.19267 [ Links ]
78. Mezey E, Imbembo AL, Potter JJ, Rent KC, Lombardo R, Holt PR. Endogenous ethanol production and hepatic disease following jejunoileal bypass for morbid obesity. Am J Clin Nutr. 1975;28(11):1277-83. https://doi.org/10.1093/ajcn/28.11.1277 [ Links ]
79. Zhao M, Zhao L, Xiong X, He Y, Huang W, Liu Z, et al. TMAVA, a Metabolite of Intestinal Microbes, Is Increased in Plasma From Patients With Liver Steatosis, Inhibits γ-Butyrobetaine Hydroxylase, and Exacerbates Fatty Liver in Mice. Gastroenterology. 2020;158(8):2266-2281.e27. https://doi.org/10.1053/j.gastro.2020.02.033 [ Links ]
80. Miura K, Ohnishi H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(23):7381-91. https://doi.org/10.3748/wjg.v20.i23.7381 [ Links ]
81. Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science. 1996;274(5290):1185-8. https://doi.org/10.1126/science.274.5290.1185 [ Links ]
82. Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113(3):414-24. https://doi.org/ 10.1172/JCI19511 [ Links ]
83. Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112(1):91-100. https://doi.org/10.1172/JCI200317797 [ Links ]
84. Musso G, Gambino R, Durazzo M, Biroli G, Carello M, Fagà E, et al. Adipokines in NASH: postprandial lipid metabolism as a link between adiponectin and liver disease. Hepatology. 2005;42(5):1175-83. https://doi.org/10.1002/hep.20896 [ Links ]
85. Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM. Adenovirus-mediated chronic «hyper-resistinemia» leads to in vivo insulin resistance in normal rats. J Clin Invest. 2004;114(2):224-31. https://doi.org/10.1172/JCI20785 [ Links ]
86. Keyhani-Nejad F, Barbosa Yanez RL, Kemper M, Schueler R, Pivovarova-Ramich O, Rudovich N, et al. Endogenously released GIP reduces and GLP-1 increases hepatic insulin extraction. Peptides. 2020;125:170231. https://doi.org/10.1016/j.peptides.2019.170231 [ Links ]
87. Dibner C, Gachon F. Circadian Dysfunction and Obesity: Is Leptin the Missing Link? Cell Metab. 2015;22(3):359-60.https://doi.org/10.1016/j.cmet.2015.08.008 [ Links ]
88. Fleet T, Stashi E, Zhu B, Rajapakshe K, Marcelo KL, Kettner NM, et al. Genetic and Environmental Models of Circadian Disruption Link SRC-2 Function to Hepatic Pathology. J Biol Rhythms. 2016;31(5):443-60. https://doi.org/10.1177/0748730416657921 [ Links ]
89. Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, et al. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell. 2016;30(6):909-924. https://doi.org/10.1016/j.ccell.2016.10.007 [ Links ]
90. Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 2004;64(20):7197-200. https://doi.org/10.1158/0008-5472.CAN-04-1459 [ Links ]
91. Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, et al. Toxicant-associated steatohepatitis. Toxicol Pathol. 2013;41(2):343-60. https://doi.org/10.1177/0192623312468517 [ Links ]
92. Aron-Wisnewsky J, Minville C, Tordjman J, Lévy P, Bouillot JL, Basdevant A, et al. Chronic intermittent hypoxia is a major trigger for non-alcoholic fatty liver disease in morbid obese. J Hepatol. 2012;56(1):225-33. https://doi.org/10.1016/j.jhep.2011.04.022 [ Links ]
93. Benotti P, Wood GC, Argyropoulos G, Pack A, Keenan BT, Gao X, et al. The impact of obstructive sleep apnea on nonalcoholic fatty liver disease in patients with severe obesity. Obesity (Silver Spring). 2016;24(4):871-7. https://doi.org/10.1002/oby.21409 [ Links ]
94. Chen J, Schenker S, Frosto TA, Henderson GI. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim Biophys Acta. 1998;1380(3):336-44. https://doi.org/10.1016/s0304-4165(98)00002-6 [ Links ]
95. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, et al. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology. 2009;50(6):1809-17. https://doi.org/10.1002/hep.23228 [ Links ]
96. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. 2009;175(4):1473-82. https://doi.org/10.2353/ajpath.2009.080999 [ Links ]
97. Sastre J, Pallardó FV, Llopis J, Furukawa T, Viña JR, Viña J. Glutathione depletion by hyperphagia-induced obesity. Life Sci. 1989;45(2):183-7. https://doi.org/10.1016/0024-3205(89)90293-2 [ Links ]
98. Baskol G, Baskol M, Kocer D. Oxidative stress and antioxidant defenses in serum of patients with non-alcoholic steatohepatitis. Clin Biochem. 2007;40(11):776-80. https://doi.org/10.1016/j.clinbiochem.2007.02.006 [ Links ]
99. Ikura Y, Ohsawa M, Suekane T, Fukushima H, Itabe H, Jomura H, et al. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: impact on disease progression. Hepatology. 2006;43(3):506-14. https://doi.org/10.1002/hep.21070 [ Links ]
100. Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, et al. Serotonin mediates oxidative stress and mitochondrial toxicity in a murine model of nonalcoholic steatohepatitis. Gastroenterology. 2007;133(2):608-18. https://doi.org/10.1053/j.gastro.2007.05.019 [ Links ]
101. Younossi ZM, Gramlich T, Bacon BR, Matteoni CA, Boparai N, O›Neill R, et al. Hepatic iron and nonalcoholic fatty liver disease. Hepatology. 1999;30(4):847-50. https://doi.org/10.1002/hep.510300407 [ Links ]
102. Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, et al. Insulin resistance-associated hepatic iron overload. Gastroenterology. 1999;117(5):1155-63. https://doi.org/10.1016/S0016-5085(99)70401-4 [ Links ]
103. Valenti L, Fracanzani AL, Bugianesi E, Dongiovanni P, Galmozzi E, Vanni E, et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology. 2010;138(3):905-12. https://doi.org/10.1053/j.gastro.2009.11.013 [ Links ]
104. Cai J, Zhang XJ, Li H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology. 2019;70(3):1026-1037. https://doi.org/10.1002/hep.30506 [ Links ]
105. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61(5):1294-303. https://doi.org/10.1007/s10620-016-4049-x [ Links ]
106. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15(6):349-364. https://doi.org/10.1038/s41575-018-0009-6 [ Links ]
107. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol Commun. 2020;4(4):478-492. https://doi.org/10.1002/hep4.1479 [ Links ]
108. Wree A, Holtmann TM, Inzaugarat ME, Feldstein AE. Novel Drivers of the Inflammatory Response in Liver Injury and Fibrosis. Semin Liver Dis. 2019;39(3):275-282. https://doi.org/10.1055/s-0039-1685515 [ Links ]
109. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898-910. https://doi.org/10.1002/hep.26592 [ Links ]
110. Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547-54. https://doi.org/10.1002/hep.27368 [ Links ]
111. Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology. 2018;155(2):443-457.e17. https://doi.org/10.1053/j.gastro.2018.04.034 [ Links ]
112. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397-411. https://doi.org/10.1038/nrgastro.2017.38 [ Links ]
113. Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7(8):425-36. https://doi.org/10.1038/nrgastro.2010.97 [ Links ]
114. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151-166. https://doi.org/10.1038/s41575-020-00372-7 [ Links ]
115. Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of Histologic Disease Activity With Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw Open. 2019;2(10):e1912565. https://doi.org/10.1001/jamanetworkopen.2019.12565 [ Links ]
116. Pais M, Franzén LE, Mathiesen UL, Kechagias S. Low clinical relevance of the nonalcoholic fatty liver disease activity score (NAS) in predicting fibrosis progression. Scand J Gastroenterol 2012;47(1):108-115. https://doi.org/10.3109/00365521.2011.634024 [ Links ]
117. Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol. 2013;59(3):550-6. https://doi.org/10.1016/j.jhep.2013.04.027 [ Links ]
118. Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, et al. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59(7):969-74. https://doi.org/10.1136/gut.2009.205088 [ Links ]
119. Argo CK, Northup PG, Al-Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol. 2009;51(2):371-9. https://doi.org/10.1016/j.jhep.2009.03.019 [ Links ]
120. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13(4):643-54.e1-9; quiz e39-40. https://doi.org/10.1016/j.cgh.2014.04.014 [ Links ]
121. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015;149(2):389-97.e10. https://doi.org/10.1053/j.gastro.2015.04.043 [ Links ]
122. Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, et al. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology. 2011;53(6):1874-82.https://doi.org/10.1002/hep.24268 [ Links ]
123. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology. 1996;24(2):289-93. https://doi.org/10.1002/hep.510240201 [ Links ]
124. Rahman RN, Ibdah JA. Nonalcoholic fatty liver disease without cirrhosis is an emergent and independent risk factor of hepatocellular carcinoma: a population based study. Hepatology. 2012;56:241A. [ Links ]
125. Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11(1):74-80. https://doi.org/10.1002/hep.1840110114 [ Links ]
126. Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, et al. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut. 2004;53(5):750-5. https://doi.org/10.1136/gut.2003.019984 [ Links ]
127. Sanyal AJ, Harrison SA, Ratziu V, Abdelmalek MF, Diehl AM, Caldwell S, et al. The Natural History of Advanced Fibrosis Due to Nonalcoholic Steatohepatitis: Data From the Simtuzumab Trials. Hepatology. 2019;70(6):1913-1927. https://doi.org/10.1002/hep.30664 [ Links ]
128. Loomba R, Adams LA. The 20% Rule of NASH Progression: The Natural History of Advanced Fibrosis and Cirrhosis Caused by NASH. Hepatology. 2019;70(6):1885-1888. https://doi.org/10.1002/hep.30946 [ Links ]
129. Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol. 2004;40(4):578-84. https://doi.org/10.1016/j.jhep.2004.02.013. [ Links ]
Citación: Prieto-Ortiz JE, Sánchez-Luque CB, Ortega-Quiroz R. Hígado graso (parte 1): aspectos generales, epidemiología, fisiopatología e historia natural. Revista. colomb. Gastroenterol. 2022;37(4):420-433. https://doi.org/10.22516/25007440.952
Recibido: 02 de Agosto de 2022; Aprobado: 23 de Septiembre de 2022
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
