










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Obstetricia y Ginecología
Print version ISSN 0034-7434On-line version ISSN 2463-0225
Rev Colomb Obstet Ginecol vol.60 no.1 Bogotá Jan./Mar. 2009
* Médico Ginecobstetra. Profesor asistente, Escuela de Medicina Universidad Industrial de Santander. Miembros del grupo de investigación GINO. Bucaramanga, Colombia. Correo electrónico: janersepulveda@yahoo.es
** Médico Ginecobstetra. Profesor titular, Escuela de Medicina Universidad Industrial de Santander. Miembros del grupo de investigación GINO. Bucaramanga, Colombia.
*** Médico Ginecobstetra. Profesor asistente, Escuela de Medicina Universidad Industrial de Santander. Miembros del grupo de investigación GINO. Bucaramanga, Colombia.
RESUMEN
Objetivo: se hace una revisión detallada de la amenorreaprimaria, teniendo como base la clasificación propuesta por Mashchak CA y col. de acuerdo con la presencia o ausencia del desarrollo mamario y la presencia o no de útero, por ser la de mayor utilidad para el enfoque de manejo de las pacientes con amenorrea primaria.
Metodología: se realizó una búsqueda de la literatura publicada en inglés a través de MEDLINE y OVID, usando como palabras clave: amenorrhea, primary amenorrhea, menstrual disorders, Turner syndrome, Kallmann syndrome, Prader-Willi síndrome, hypogonadotropic-hypogonadism; y se clasificó la información como soporte de la presente revisión, realizando resúmenes para su análisis.
Resultados: la amenorrea primaria puede ser causada por una variedad de alteraciones que incluyen anormalidades müllerianas, gonadales, hipofisiarias, hipotalámicas, adrenales y tiroideas, o disfunciones hormonales en estos diferentes niveles. Estas anormalidades pueden ser congénitas por defectos cromosómicos o genéticos, o adquiridas, por lo tanto, es importante realizar un diagnóstico certero de esta patología para llevar a cabo un enfoque terapéutico adecuado, con el fin de disminuir todas las consecuencias que la enfermedad puede causar. Conclusiones: el tratamiento de las pacientes con amenorrea primaria debe ser individualizado, de acuerdo con las posibilidades terapéuticas de cada paciente, pero existen unas preguntas generales que tienen todas las pacientes o sus familiares y son relacionadas con la menstruación y los ciclos menstruales espontáneos posteriores, fertilidad, sexualidad y posibilidad de coitos con penetración vaginal satisfactoria.
Palabras clave: amenorrea, desórdenes menstruales, síndrome de Turner, síndrome de Kallmann, síndrome de Prader-Willi, hipogonadismo hipogonadotrópico.
SUMMARY
Objective: this is a detailed review of primary amenorrhea using Mashchak CA et al. classification according to the presence or absence of breast development and the presence or absence of uterus as being the most useful approach for managing patients suffering from this problem.
Method: Medline and Ovid databases were searched for papers published in English using the following keywords: amenorrhea, primary amenorrhea, menstrual disorder, Turner syndrome, Kallmann syndrome, Prader-Willisyndrome, hypogonadotropic hypogonadism. This information was classified to support this review by making summaries for analysis.
Results: primary amenorrhea can be caused by many alterations affecting the Mullerian structures, gonads, pituitary gland, hypothalamus, thyroid, adrenals or hormonal dysfunction; such anomalies may be congenital due to genetic or chromosomal defects or acquired. It is thus important that this problem is specifically diagnosed to enable a suitable therapeutic approach to be adopted for minimising the consequences of this disease.
Conclusions: many diseases cause this problem, so diagnosing and treating patients suffering from primary amenorrhea must be individualised; however, some general questions needing specific answers are raised by all patients or their families. These questions are related to menstruation and spontaneous menstrual cycles, subsequent fertility, sexuality and the possibility of coitus with satisfactory vaginal penetration.
Key words: amenorrhea, primary amenorrhea, menstrual disorder, Turner syndrome, Kallmann syndrome, Prader-Willi syndrome, hypogonadotropic-hypogonadism.
INTRODUCCIÓN
Se define como amenorrea primaria la ausencia de menarquia a los 16 años; a los 14 años, si hay ausencia de pubarquia; o a los 13 años, si hay ausencia de telarquia;1,2 también recibe este nombre cuando transcurren más de 5 años entre la telarquia y la menarquia.1 Un criterio de inclusión para identificar estas pacientes es que los genitales externos sean fenotípicamente femeninos; de lo contrario, se estudia como una ambigüedad sexual.
Se considera que la incidencia de amenorrea primaria es menor al 0,1%.3
La amenorrea no es un síndrome ni un diagnóstico, es la manifestación de múltiples causas. En algunos períodos de la mujer son fisiológicas, como en la infancia, el embarazo, el puerperio, la lactancia y la menopausia. Sin embargo, si se presenta en otras circunstancias durante el período reproductivo, se consideran patológicas. De acuerdo con su aparición puede clasificarse en primaria o secundaria, según la edad de la menarquia.3
La amenorrea primaria puede ser causada por una variedad de desórdenes que incluyen anormalidades genéticas, alteraciones müllerianas, lesiones hipotálamo-hipofisiarias y disfunción hormonal.4 El diagnóstico certero de esta patología favorece un enfoque terapéutico adecuado, con el fin de atenuar o eliminar las consecuencias de esta perturbación,3,5 ya que un tratamiento tardío puede ocasionar alteraciones en el desarrollo de la personalidad o en la salud de la adolescente.5 Por lo tanto, el objetivo de este documento es hacer una revisión de la etiología de la amenorrea primaria, de acuerdo con el desarrollo mamario y la presencia o no de útero, por ser de utilidad para el manejo de estas pacientes.
METODOLOGÍA
Se realizó una búsqueda de la literatura publicada en inglés, a través de MEDLINE y OVID, usando como palabras clave: amenorrhea, primary amenorrhea, menstrual disorders, Turner syndrome, Kallmann syndrome, Prader-Willi syndrome, hypogonadotropic-hypogonadism; y se clasificó la información como soporte de la presente revisión, realizando resúmenes para su análisis.
Clasificación
Existe una clasificación de la amenorrea primaria de acuerdo con la causa:5
• Congénitas: son la causa más frecuente, se deben a alteraciones cromosómicas o genéticas, produciendo alteraciones enzimáticas o de receptores.
• Adquiridas: pueden ser funcionales u orgánicas.
• Anatómicas: cuando la causa está localizada en el órgano efector (útero y endometrio) o en las vías de drenaje (vulva y vagina). Puede ser congénita o adquirida.
• Endocrinas: se deben a una alteración funcional u orgánica en el eje del hipotálamo, hipófisis y ovario.
Hay otras clasificaciones como la división en amenorreas por compartimientos: origen central (hipotálamo e hipófisis) o periférica que puede ser ovárica, del órgano efector2 y del tracto de salida o vagina; de origen gonadal o extragonadal o de acuerdo con la secreción de gonadotropinas (hiper, hipo o normogonadotropa).5
Mashchak CA y col.4 clasificaron en 1981 la amenorrea primaria de una forma clínica en cuatro categorías, de acuerdo con la presencia o ausencia de desarrollo mamario y de la presencia o no de útero. A continuación se hace una descripción de las diferentes causas, teniendo en cuenta esta clasificación por ser la más práctica y la de mayor utilidad para su estudio.
1. CATEGORÍA I
Ausencia de mamas y presencia de útero
Es un grupo complejo para su análisis por presentar múltiples entidades que pueden llevar a amenorrea primaria. Es el segundo en frecuencia. Se puede clasificar en 3 subgrupos:
1.1. Falla hipotalámica: causada por la secreción inadecuada de la hormona liberadora de gonadotropina (GnRH). Hay un hipogonadismo hipogonadotrópico hipotalámico con bajos niveles de gonadotropinas, pero cuando se estimula con GnRH éstas suben, demostrando que la hipófisis es normal.3 Las principales causas de este grupo son:
a. Secreción insuficiente de neurotransmisores como dopamina y norepinefrina.
b. Síntesis inadecuada de GnRH idiopática asociada a talasemia mayor, la cual se diferencia del síndrome de Kallmann porque no cursa con anosmia.6
c. Defectos anatómicos congénitos.
d. Ausencia del piso de la silla turca con encefalocele anterior.
e. Neoplasias como el craniofaringioma, el cual puede presentarse con retardo en el crecimiento, defectos en los campos visuales y diabetes insípida;6 se considera que es un tumor extracelular que interfiere con la síntesis y secreción de GnRH o la estimulación de las gonadotropinas hipofisiarias.7 El pico de incidencia es entre los 6 y 14 años. Las imágenes revelan una silla turca anormal y calcificaciones en el 70% de los casos.2
f. Síndrome de Kallmann: con una incidencia de 1 en 10.000 a 1 en 86.000 nacidos vivos;6 en el cual el hipogonadismo hipogonadotrópico está asociado con anosmia y agenesia del bulbo olfatorio. Este camino común de migración de GnRH y neuronas olfatorias es interrumpido, se han encontrado defectos génicos en las proteínas que facilitan esta migración neuronal. El defecto génico que ocasiona pérdida de esta proteína adherente facilitadora se ha localizado en el cromosoma X en la forma del síndrome ligado a X, y este locus se ha designado como KALI G-1 (gen 1 de intervalo de síndrome de Kallmann),8,9 el cual es detectado desde la semana 11 de gestación. Los hallazgos en las imágenes radiográficas del hipotálamo y de la hipófisis son normales, pero la ausencia o anormalidad en la morfología del bulbo olfatorio en la resonancia nuclear magnética permite un diagnóstico presuntivo de este síndrome.6 Otros investigadores han demostrado un bulbo olfatorio normal en el 25% de pacientes con el síndrome de Kallmann, lo cual sugiere que esta técnica no es lo suficientemente sensible para diferenciar el síndrome de Kallmann de la síntesis inadecuada de GnRH idiomática.10 Además de la anosmia, una variedad de anomalías que pueden ocurrir en el síndrome de Kallmann han sido reportadas, incluyendo paladar hendido y labio hendido, disquinesia, ataxia, sordera nerviosa, cerebelosa, agenesia renal unilateral y anomalías del mecanismo de la sed y descarga de vasopresina.8-12 Han sido documentadas tres formas de transmisión: ligado al cromosoma X, autosómico dominante y autosómico recesivo,2 sin embargo, las dos terceras partes de los casos son esporádicos.13
g. Síndrome de Prader-Labhart-Willi: este síndrome ocurre cuando hay una delección en el brazo largo del cromosoma 1514-17 con una ocurrencia de 1-10.000 a 1-15.000.14 Las manifestaciones aparecen desde la vida prenatal, en la cual hay hipotonía que causa disminución de movimientos a. Síndrome de Turner: es el más frecuente de este fetales, posición fetal anormal y dificultades en grupo.3 La frecuencia de este síndrome en la el momento del parto, a menudo ocasionando concepción es aproximadamente del 3%, pero el necesidad de cesárea.14 Posteriormente en la 99% son abortados espontáneamente; tiene una vida neonatal esa hipotonía está asociada con frecuencia de 1-2000 a 1-7000 nacidos vivos, con pobre succión, que puede resultar en problemas de crecimiento.18 En la vida infantil se manifiesta con letargia14 y los reflejos pueden estar ausentes o disminuidos. En la vida adulta persiste la hipotonía con disminución en el tono y masa muscular, baja estatura, retardo mental, manos y pies pequeños y tendencia a la obesidad;8,11,14,18-21 también puede presentarse osteoporosis, estrabismo y a menudo escoliosis y/o en herradura) y desórdenes autoinmunes como cifosis,19 hipoplasia de labios menores y clítoris; se considera que hay una alteración en el núcleo paraventricular del hipotálamo, que es la causa de la obesidad en estos pacientes.19,22 La mayoría de los individuos con este síndrome tienen retardo mental en un 97%.18 Aproximadamente del de 5 a 10% de los pacientes pueden presentar problemas psiquiátricos.14
h. Síndrome Laurence-Moon-Biedl: se caracteriza por talla baja, retinitis pigmentosa, sordera, paraplejía espástica, polidactilia, sindactilia, obesidad, hipogonadismo y retardo mental.5,8
i. Síndrome de Frolich: este síndrome cursa con talla baja, obesidad y retardo mental leve, ocasionado por una alteración del área ventromedial del hipotálamo de carácter tumoral.5
1.2. Falla hipofisiaria: estas pacientes no tienen una adecuada respuesta de secreción de LH y FSH al realizar estimulación con GnRH.3 En este grupo se identifican:
a. Insuficiencia aislada de gonadotrofinas que pueden ser heredadas con un gen autonómico dominante, autosómico recesivo o rasgo ligado al cromosoma X.11
b. Encefalitis.
c. Hipotiroidismo prepuberal.
1.3. Falla gonadal: las pacientes de este grupo tienen hipogonadismo hipergonadotrópico.3 A este grupo pertenecen:
a. Síndrome de Turner: es el más frecuente de este grupo.3 La frecuencia de este síndrome en la concepción es aproximadamente del 3%, pero el 99% son abortados espontáneamente; tiene una frecuencia de 1-2000 a 1-7000 nacidos vivos, con un promedio de 1-2500;8,23 y se caracteriza por ausencia de uno de los cromosomas X.24 Las características clínicas asociadas con este síndrome son baja estatura, anomalías somáticas, cuello membranoso, paladar ojival, micrognatia, enfermedad cardíaca congénita (con mayor frecuencia coartación de la aorta), uñas hiperconvexas en las manos, anomalías renales (más a menudo riñón en herradura) y desórdenes autoinmunes como la tiroiditis y la enfermedad de Addison.8,24,25 Se ha reportado menstruación espontánea en 2 a 5% de las niñas con síndrome de Turner.2 La incidencia de gonadoblastomas es aproximadamente del 12%.23
b. Síndrome de Bonnevie-Ullrich: variante del síndrome de Turner pero con talla normal.
c. Síndrome de Noonan: el cariotipo de estas pacientes es 46, XX; es un desorden autonómico dominante, que fue descrito por primera vez en 1963.26-28 Se estima que su incidencia es de 1-2500 nacidos vivos,29 está caracterizado por baja estatura, enfermedades cardíacas congénitas, genitales externos hipoplásicos y facies distintivas, incluyendo hipertelorismo, fisuras palpebrales, epicanto y orejas de implantación baja.26,27 Se han reportado casos de leucemia mielomonocítica crónica, asociada con este síndrome.29
d. Síndrome de Vancouver Devriendt: es autonómico recesivo, se caracteriza por baja estatura, retardo mental, dimorfismo facial, alopecia, ataxia, extrapiramidalismo y disartria de aparición en la adolescencia con amenorrea primaria.
e. Anomalías estructurales del cromosoma X: el cariotipo de este desorden es 46, XX, pero uno de los cromosomas X es incompleto, la deleción puede ser del brazo largo o corto del cromosoma X. Pacientes con deleción del brazo largo del cromosoma largo usualmente tienen estatura normal, no presentan anormalidades somáticas, pueden tener bandas gonadales, infantilismo sexual y esterilidad.3 Las pacientes con deleción del brazo corto del cromosoma usualmente tienen un fenotipo similar al síndrome de Turner.3
f. Mosaicismo: el más común es X/XX, el 80% de las pacientes cursan con baja estatura y el 66% tienen anomalías somáticas. Aproximadamente el 20% de estas pacientes tienen menstruación espontánea3 y puede presentarse embarazo.13 Los hallazgos clínicos en el mosaicismo X/XXX y X/XX/XXX son similares al anterior.
g. Disgenesia gonadal pura: este término se refiere a las mujeres cuyo cariotipo es 46, XX o 46, XY y tienen bandas gonadales. Este trastorno puede ocurrir esporádicamente o heredarse como rasgo autosómico recesivo o ligado al cromosoma X, en caso de digenesia gonadal XY.8 Estas pacientes tienen características sexuales infantiles, estatura normal y no presentan anomalías somáticas; sin embargo, se han reportado casos de características sexuales secundarias normales y una historia de hemorragia espontánea.3 Cuando ocurre disgenesia gonadal en individuos 46, XY, se denomina síndrome de Swyer-James,8,30 fue descrito por primera vez en 1955 y se han reportado 62 pacientes en la literatura hasta 1994.30 Su etiología aún permanece desconocida. La malignización de la gónada fibrosa en este síndrome ocurre en un 10 a 30% de las pacientes; por esta razón está indicada claramente la gonadectomía para prevenir el desarrollo de neoplasia de las células germinales. Los tumores más frecuentes son el gonadoblastoma en el 75% de los casos y el disgerminoma.31 Se ha reportado en la literatura un caso de invasión intestinal de un disgerminoma de un paciente con este síndrome.32
h. Deficiencia de 17-hidroxilasa con cariotipo 46, XX: esta enzima es necesaria para la conversión de pregnenolona en 17-hidroxipregnenolona y de progesterona en 17-hidroxiprogesterona, esta acción enzimática está codificada en el cromosoma 10.33,34 Esta alteración cursa con disminución de cortisol, que causa aumento de ACTH y aumento en la producción de mineralocorticoides, ocasionando retención de sodio, hipertensión y pérdida de potasio.3,7 Estas pacientes presentan bajo nivel de 17-alfa hidroxiprogesterona menor de 0,2 ng/mL, elevación de las concentraciones séricas de progesterona (>3ng/mL) y concentración elevada de desoxicorticosterona. Después de la administración de ACTH hay un aumento en las concentraciones de progesterona y ningún cambio en el nivel de 17-alfa hidroxiprogesterona.7
Diagnóstico de las pacientes en la categoría I
El diagnóstico de este grupo se puede enfocar desde el punto de vista clínico de acuerdo con la talla, presencia de retardo mental y de obesidad (figura 1 y 2).
El estudio inicial de este grupo se realiza solicitando FSH,3,4 se subdividen las pacientes en hipergonadotrópicas con FSH alta, mayor de 20 mUI/mL, y normo u hipogonadotrópicas con FSH normal o baja, menor de 10 mUI/mL.
2. CATEGORÍA II
Mamas presentes y útero ausente
A este grupo pertenecen:
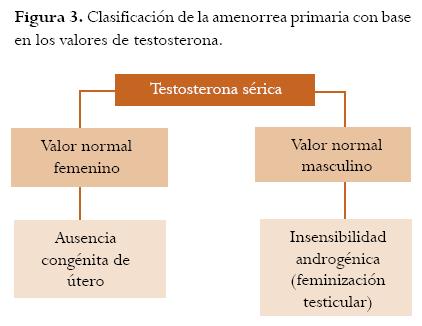
2.1. La insensibilidad androgénica (feminización testicular completa): estas pacientes tienen un cariotipo 46, XY, testículos y fenotipo femenino, ausencia de vello púbico y axilar; está ligado al cromosoma X recesivo o dominante con penetración incompleta3 y se presenta carencia de receptores androgénicos o defectos en el funcionamiento normal, presentes en el citosol o núcleo de las células blanco. Los ovarios, útero y trompas están ausentes, porque los testículos secretan normalmente factor inhibidor mülleriano (MIF), el cual no requiere receptores para su acción.2,3,7,24 Estas personas suelen ser altas y manifiestan tendencia eunucoide (brazos largos con manos y pies grandes,7 las mamas son voluminosas). El testículo produce normalmente testosterona y estradiol, por lo tanto en las gonadotropinas usualmente hay aumento de LH con FSH normal. El estradiol está en niveles bajos (<30 pg/mL) debido a la oposición androgénica; la pequeña cantidad de estrógenos secretados por los testículos y adrenales son producidos por la conversión periférica de androstenodiona a estrona y estradiol. El nivel de testosterona está en nivel masculino (>3ng/dl).3 Estas pacientes también requieren remoción gonadal por el aumento de malignidad, aunque ésta se presenta más tardíamente, al contrario de la digenesia gonadal pura XY y su incidencia es sólo del 5 a 10%.2
2.2. Ausencia congénita de útero, los ovarios están presentes, la vagina está ausente (síndrome de Mayer-Rokitansky-Küster-Hauser): su incidencia es de 1 en 4000 nacimientos femeninos,2 puede estar asociada con anomalías urinarias, mayores en un 15% (ausencia de riñón) y menores en un 40% (sistema colector doble).1,3,7 También puede haber anomalías óseas hasta en un 5% (la fusión de vértebras cervicales es la más frecuente) o anomalías cardíacas.
Diagnóstico de las pacientes en la categoría II
El estudio de este grupo se inicia con testosterona sérica (figura 3).
3. CATEGORÍA III
Mamas ausentes y útero ausente
Es el grupo menos frecuente, el cariotipo es masculino y los niveles de gonadotropinas son elevados. La testosterona está normal o baja en el rango femenino.3 A este grupo pertenecen:
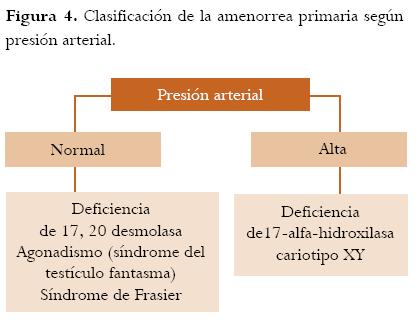
3.1. Deficiencia de 17,20 desmolasa: es la enzima responsable de convertir la 17-alfa-hidroxipregnenolona a dehidroepiandrosterona y la 17-hidroxiprogesterona a androstenodiona. Los testículos deben ser removidos por el riesgo de malignización.3 Las concentraciones de cortisol reaccionan normalmente a la estimulación de ACTH. Las concentraciones de testosterona son bajas.7
3.2. Agonadismo (síndrome del testículo fantasma): estos pacientes no presentan órganos sexuales internos y no tienen trastorno de la esteroidogénesis; como no hay presencia de testículos, no tienen foco para secreción de MIF. Se ha postulado que en la época embrionaria desarrollan tejido testicular y el MIF suprime el desarrollo mülleriano, pero posteriormente el testículo desaparece, por lo cual se denomina síndrome del testículo fantasma.3
3.3. Deficiencia de 17-alfa-hidroxilasa con cariotipo XY: estas pacientes presentan hipertensión pero no presentan útero, a diferencia de quienes tienen deficiencia de 17-alfa-hidroxilasa con cariotipo XX.
3.4. Síndrome de Frasier: caracterizado por neuropatía progresiva, infantilismo sexual, falla renal y gónadas rudimentarias en las cuales se puede desarrollar gonadoblastomas. Es causado por una mutación en el gen WT1.11
Diagnóstico de las pacientes en la categoría III
El estudio inicial es con cariotipo. Clínicamente también se puede estudiar este grupo de acuerdo a la presencia o no de hipertensión (figura 4).
4. CATEGORÍA IV
Mamas presentes y útero presente
Es el grupo más frecuente y comprende entre el 30 y el 40% de los casos de amenorrea primaria. El 25% de este grupo presenta galactorrea, prolactina elevada y tomografía anormal de la silla turca compatible con adenoma hipofisiario.3
El otro 75% tiene prolactina normal e incluye pacientes con síndrome de ovario poliquístico, hiperplasia suprarrenal, disfunción hipotalámica, falla hipotálamo-hipofisiaria y falla ovárica por ooforitis autoinmune, por quimioterapia o radioterapia.13
Recientemente se han reportado casos de amenorrea primaria por mutación a nivel del receptor de la hormona luteinizante (LH) y bajos niveles de estradiol.35,36 En estas pacientes se presentan cambios a nivel del receptor, ya sea por sustitución de lisina por ácido glutámico en la posición 354 del receptor de LH34 o de alanina por prolina en la posición 593 del receptor de LH.36
Finalmente, en este grupo debe descartarse la falsa amenorrea o criptomenorrea debida a la obstrucción del tracto de salida con acumulación del fluido menstrual, causado por agenesia cervical, himen imperforado o septum vaginal transverso. Estas entidades se caracterizan por dolor cíclico severo e incapacitante en niñas con desarrollo puberal normal; al realizar ecografía pélvica se encuentra imagen de líquido acumulado en la vagina, útero o trompas.5
Diagnóstico de las pacientes en la categoría IV
El estudio inicial de estas pacientes se realiza solicitando niveles de prolactina y hormona tiroestimulante (TSH).
ENFOQUE DE MANEJO
El interrogatorio para las pacientes con amenorrea primaria
Las preguntas deben tener una adecuada secuencia para identificar la causa de la amenorrea primaria:13
En la anamnesis se debe hacer énfasis en:
• El desarrollo de las mamas y caracteres sexuales secundarios que sugieran falla ovárica o hipofisiaria, o anormalidades cromosómicas.
• Historia familiar que sugiera un desorden familiar, haciendo énfasis en trastornos mentales.
• Estatura en mujeres miembros de la familia, que puedan indicar síndrome de Turner o enfermedad hipotalámica-hipofisiaria.
• Enfermedades neurológicas en la infancia.
• Síntomas de virilización: la presencia de virilización sugiere síndrome de ovario poliquístico, tumores de ovario o adrenales que liberan andrógenos, o presencia de material cromosómico.
• Historia de estrés, cambio de peso, dieta o ejercicio que pueda resultar en amenorrea hipotalámica.
• Ingesta de medicamentos que puedan causar amenorrea.
• Presencia de galactorrea por exceso de prolactina.
• Síntomas de enfermedades hipotálamohipofisarias, incluyendo cefalea, visión borrosa, poliuria y polidipsia.
El examen físico
Se debe hacer énfasis en:
• Evaluación del desarrollo puberal: caracteres sexuales secundarios.
• Antropometría: estatura, peso, cintura, cadera, índice de masa corporal, relación de segmentos superior-inferior y relación de talla-envergadura.
• Coeficiente intelectual.
• Palpación de la tiroides.
• Desarrollo mamario.
• Examen cuidadoso de los genitales, tamaño del clítoris, aspecto de labios mayores y menores, orificio himeneal.
• Examen de la piel en búsqueda de hirsutismo, acné, estría, hiperpigmentación o vitíligo.
Ayudas diagnósticas
Es necesaria la ecografía para identificar la presencia o ausencia de útero. Se deben realizar exámenes de laboratorio individualizados, los cuales deben ser solicitados de acuerdo con la clasificación propuesta de Mashchak CA y colaboradores.
Finalmente, después de realizar la anamnesis, el examen físico y solicitar las pruebas de laboratorio, se deben individualizar los manejos para cada entidad; sin embargo, hay unas preguntas generales que tienen todas las pacientes o sus familiares y unas reglas fundamentales de manejo que se deben responder y aplicar a todas las pacientes:37
• Menstruación y ciclos menstruales espontáneos posteriores.
• Fertilidad.
• Sexualidad y posibilidad de coitos con penetración vaginal satisfactoria.
Recomendaciones para el manejo de estas pacientes
Son pertinentes las siguientes recomendaciones:
a. Debe evitarse el uso de términos como pseudohermafroditismo, hermafroditismo, testículo feminizante, cariotipo XY y en general todas las expresiones que lleven a la paciente o sus familiares a pensar que es de género masculino. Se les debe decir que su desarrollo fue incompleto antes o después del nacimiento y por ello están presentando el problema actual, para evitar todos los conflictos psicológicos que se podrían presentar en una paciente con un rol de género femenino bien establecido.37
b. No realizar tacto rectal para aclarar la presencia de útero en la primera consulta, porque en muchas ocasiones no aclara la situación y es muy agresivo psicológicamente para la paciente. La presencia de útero se establece en la actualidad con la ecografía pélvica por vía abdominal, que está fácilmente disponible en muchas instituciones, inclusive de primer nivel.37
c. No realizar laparoscopia diagnóstica. De acuerdo con la clasificación propuesta no se ve la necesidad en ninguna de las entidades descritas.37
d. Cuando se tenga claro el diagnóstico, se realiza laparoscopia terapéutica solamente para extirpar la gónada que tenga posibilidad alta de desarrollo tumoral maligno por la presencia del cromosoma y/o el antígeno H-Y.37
e. Para crear una cavidad vaginal en las pacientes que tienen agenesia vaginal, como en el síndrome de Rokitansky, y las de insensibilidad androgénica, se debe hacer tratamiento inicialmente con el uso de dilatadores. Si este método no es exitoso se recurre a la cirugía con injertos de piel de región glútea o con intestino.37-40
f. Hay que dar suplementos hormonales inicialmente con estrógenos a dosis bajas para estimular el desarrollo mamario en quienes no han tenido la telarquia. Cuando pasan seis meses de tratamiento sin incremento en el volumen mamario, se adicionan los progestágenos para hacer ciclos bifásicos en las pacientes que tienen útero, logrando así los sangrados cíclicos y la prevención de la osteoporosis y otras enfermedades derivadas de la ausencia de los estrógenos.37
g. A las pacientes que tienen ovarios normales y útero, como el síndrome de Kallman y el déficit aislado de GnRh, y desean quedar embarazadas, se les hace la inducción de la ovulación con la bomba de GnRh en los países donde está disponible o se hace la inducción con FSH recombinante o gonadotropinas menopáusicas humanas y gonadotropina coriónica.37
h. Las pacientes con disgenesia gonadal y útero pueden recurrir a la donación de oocitos para lograr un embarazo.37
i. Las afectadas por aplasia o hipoplasia uterina pueden recurrir al préstamo de útero o maternidad subrogada y lograr un hijo o hija biológicos, pero sin maternidad gestacional, porque sus ovarios son normales.37
j. Deben recibir asesoría y orientación psicológica.13
k. Tratamiento precoz del hiperandrogenismo para prevenir consecuencias a largo plazo, como la hiperplasia endometrial, obesidad y defectos metabólicos.13
l. En caso de amenorrea hipotalámica funcional, realizar control de ganancia de peso, reducción en la intensidad de ejercicio o resolución de problemas que lleven a estrés.13
m. En pacientes con síndrome de Turner está indicada la hormona de crecimiento recombinante y la suplencia hormonal.23,41
REFERENCIAS
1. Pletcher JR, Slap GB. Menstrual disorders. Amenorrhea. Pediatr Clin North Am 1999;46:505-18 [ Links ]
2. Speroff L, Glass RH, Kase NG. Clinical ginecologic endocrinology and infertility. 6th ed. Baltimore, Md.: Lippincott Williams & Wilkins; 1999. [ Links ]
3. Davajan V, Kletzky OA. Primary Amenorrhea. Phenotypic Female External Genitalia. En: Mishell D, Davajan V, Lobo R. Infertility, Contraception & Reproductive Endocrinology. 3rd. ed. Boston: Blackwell Scientific Publications; 1991. p. 356-71. [ Links ]
4. Mashchak AC, Kletzky OA, Davajan V, Mishell DR Jr. Clinical and laboratory evaluation of patients with primary amenorrhea. Obstet Gynecol 1981;57:715-21. [ Links ]
5. Sánchez Escobar F, García Gómez JE. Amenorrea primaria. En: Botero J, Jubiz A, Henao G. Obstetricia y Ginecología. 7a. ed. Universidad de Antioquia, Colombia: Quebecor Impreandes; 2004. p. 492-502. [ Links ]
6. Hayes FJ, Seminara SB, Crowley WF Jr. Hypogonadotropic hypogonadism. Endocrinology and Metabolism Clinics North Am 1998;27:739-63. [ Links ]
7. Scherzer WJ, McClamrock H. Amenorrea. En: Berek J, Hillard PA, Adashi EY. Ginecología de Novak. 12 ed. México DF: McGraw-Hill. Interamericana; 1997. p. 809-32. [ Links ]
8. Berek J, Hillard PA, Adashi EY, Rebar RV. Pubertad. En: Ginecología de Novak. 12 ed. México DF: McGraw-Hill Interamericana; 1997. p. 783. [ Links ]
9. Hay C, Wu F. Genetics and hypogonadotrophic hypogonadism. Curr Opin Obstet Gynecol 2002;14:303-8. [ Links ]
10. Quinton R, Duke VM, de Zoysa PA, Platts AD, Valentine A, Kendall B, et al. The neuroradiology of Kallmann’s syndrome: a genotypic and phenotypic analysis. J Clin Endocrinol Metab 1996;81:3010-7. [ Links ]
11. OMIM® -Online Mendelian Inheritance in Man®. Visitado 2008, Abr 8. Disponible en: http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim [ Links ]
12. Waldstreicher J, Seminara SB, Jameson JL, Geyer A, Nachtigall LB, Boepple PA, et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab 1996;81:4388-95. [ Links ]
13. Welt CK, Barbieri RL. Etiology, diagnosis, and treatment of primary amenorrhea. UpToDate 2007;15(3). [ Links ]
14. Cassidy SB, Schwartz S. Prader-Willi and Angelman syndromes. Disorders of genomic imprinting. Medicine (Baltimore) 1998;77:140-51. [ Links ]
15. Holm VA, Cassidy SB, Butler MG, Hanchett JM, Creenswag LR, Whitman By, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 1993;91:398-402. [ Links ]
16. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenen SB, Crawford JK. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med 1981;304:325-9. [ Links ]
17. Festen D, Wevers M, de Weerd AW, van den Bossche RA, Duivenvoorden HJ, Otten BJ, et al. Psychomotor development in infants with prader-willi syndrome and associations with sleep-related breathing disorders. Pediatr Res 2007;62:221-4. [ Links ]
18. Miller SP, Riley P, Shevell MI. The neonatal presentation of Prader-Willi syndrome revisited. J Pediatr 1999;134:226-8. [ Links ]
19. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Endocrinol Metab 1995;80:573-9. [ Links ]
20. Belt AB, Hertel TA, Mante JR, Marks T, Rockett VL, Wade C, et al. Movement Characteristics of Persons with Prader-Willi Syndrome Rising from Supine. Pediatr Phys Ther 2001;13:110-21. [ Links ]
21. McEntagart ME, Webb T, Hardy C, King MD. Familial Prader-Willi syndrome: case report and a literature review. Clin Genet 2000;58:216-23. [ Links ]
22. Smeets DF, Hamel BC, Nelen MR, Smeets HJ, Bollen JH, Smits AP et al. Prader-Willi syndrome and Angelman syndrome in cousins from a family with a translocation between chromosomes 6 and 15. N Engl J Med 1992;326:807-11. [ Links ]
23. Stratakis CA, Rennert OM. Turner syndrome: an update. Endocrinologist 2005;15:27-36. [ Links ]
24. Braverman PK, Sondheimer SJ. Menstrual disorders. Pediatr Rev 1997;18:17-25. [ Links ]
25. Sybert VP, MsCauley E. Turner´s syndrome. N Engl J Med 2004;351:1227-38. [ Links ]
26. Bader-Meunier B, Tchernia G, Mielot F, Fontaine JL, Thomas C, Lyonnet S, et al. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr 1997;130:885-9. [ Links ]
27. Silburn PA, Nicholson GA, Teh BT, Blair IP, Pollard JD, Nolan PJ, et al. Charcot-Marie-Tooth disease and Noonan syndrome with giant proximal nerve hypertrophy. Neurology 1998;50:1067-73. [ Links ]
28. Yoshida R, Hasegawa T, Hasegawa Y, Nagai T, Kinoshita E, Tanaka Y, et al. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan syndrome. J Clin Endocrinol Metab 2004;89:3359-64. [ Links ]
29. Romano AA, Blethen SL, Dana K, Noto RA. Growth hormona treatment in Noonan syndrome: the National Cooperative Growth Study experience. J Pediatr 1996;128:S18-21. [ Links ]
30. Villegas JD, Cuevas R, Barón G. Disgenesia gonadal pura: síndrome de Swyer-James. Presentación de un caso y revisión de la literatura. Rev Colomb Obstet Ginecol 1998;49:41-5. [ Links ]
31. Wilson EE, Vuitch F, Carr BR. Laparoscopic removal of dysgenetic gonads containing a gonadoblastoma in a patient with Swyer syndrome. Obstet Gynecol 1992;79:842-3. [ Links ]
32. Parker M, Barnhill D, Teneriello M, O’Connor D, Park R. Intestinal invasion by a dysgerminoma in a patient with Swyer syndrome. Obstet Gynecol 1992;80:567-8. [ Links ]
33. Miller WL. Pathophysiology, genetics, and treatment of hyperandrogenism. Pediatr Clin North Am 1997;44:375-95. [ Links ]
34. Fardella CE, Hum DW, Homoki J. Miller WL. Point mutation of Arg440 to his cytochrome P450c 17 causes severe 17 alfa hydroxylase deficiency. J Clin Endocrinol Metab 1994:79:160-4. [ Links ]
35. Stavrou SS, Zhu YS, Cai LQ, Katz MK, Herrera C, Defillo-Ricart M, et al. A novel mutation of the human luteinizing hormone receptor in 46 XX sisters. J Clin Endocinol Metab 1998;83:2091-8. [ Links ]
36. Toledo SP, Brunner HG, Kraaij R, Post M, Dahia PL, Hayashida CY, et al. An inactivating mutation of the luteinizing hormone receptor causes amenorrhea in a 46, XX female. J Clin Endocrinol Metab 1996;81:3850-4. [ Links ]
37. Jaimes H. Amenorrea primaria. En: Pérez LE. Infertilidad y Endocrinología Reproductiva. 3a ed. Bogotá: Grafiempresos Donado; 2007. p. 199-210. [ Links ]
38. Stany MP, Winter WE 3rd, Elkas JC, Rose GS. The uses of a cellular termal grafo for vulvo vaginal reconstruction in a patient with lichen planus. Obstet and Gynecol 2005;105:1268-71. [ Links ]
39. Wierrani F, Grûnberger W.Vaginoplasty using deepthelialized vulvar transposition Flaps: the Grûberger method. J Am Coll Surg 2003;196:159-62. [ Links ]
40. Motoyama S, Laoag-Fernández JB, Mochizuki S, Yamabe S, Maruo T. Vaginoplasty with intercede absorbable adhesion barrier for complete squamous epithelization in vaginal agenesis. Am J Obstet Gynecol 2003;188:1260-4. [ Links ]
41. Stephure DK. The Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in turner syndrome: results of the Canadian Randomized controlled trial. J Clin Endocrinol Metab 2005;90:3360-6. [ Links ]
Conflicto de intereses: ninguno declarado.

