











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El síndrome de encefalopatía mitocondrial, acidosis láctica y episodios similares a un accidente cerebrovascular (MELAS, por su sigla en inglés) forma parte de las enfermedades mitocondriales y los desórdenes hereditarios que se manifiestan como consecuencia de la deficiencia de una o varias proteínas a nivel mitocondrial. El síndrome MELAS causa compromiso multiorgánico y afecta principalmente las células cerebrales. La mutación encontrada con mayor frecuencia (80% de los casos) en este trastorno es la A3243G del gen MT-TL1 en el ADN mitocondrial (ADNmt), que codifica para el ARN de trasferencia (ARNt).1,2
Las manifestaciones clínicas del síndrome MELAS pueden tardar varios años en aparecer, lo cual dificulta el diagnóstico temprano. En el 62% de los pacientes la enfermedad se desarrolla antes de los 15 años;3 sin embargo, ocasionalmente puede presentarse de manera temprana, tal como se evidencia en el caso descrito a continuación.
Presentación del caso
Paciente femenina de cinco años de edad quien fue llevada a consulta al servicio de pediatría por cuadro de tres meses de evolución de ataxia progresiva que le dificultaba la marcha. Previamente había presentado dos episodios convulsivos parciales complejos interpretados como epilepsia focal. Como antecedente se reportó epilepsia en un tío materno fallecido a los 12 años; los padres no eran consanguíneos. Aunque al ingreso los laboratorios fueron normales, se observó hiperlactatemia. La tomografía axial computarizada (TAC) de cráneo y el electroencefalograma fueron normales, mientras que la resonancia magnética nuclear (RMN) cerebral evidenció una leve acentuación de los surcos corticales en ambos hemisferios cerebrales y una leve atrofia cerebelosa. Se indicó ácido valproico más lamotrigina. Debido a la persistencia de lactato sérico elevado, se solicitó relación lactato/piruvato en sangre y el resultado fue >20, por lo que se sospechó enfermedad mitocondrial.
Seis meses después, la paciente presentó deterioro neurológico progresivo, disminución de la fuerza muscular, persistencia de la marcha atáxica, mutismo y falta de seguimiento visual. La TAC de cráneo de control evidenció lesión sugestiva de infarto cerebral en el lóbulo occipital derecho y la RMN en T1 mostró hipointensidad cortico-subcortical temporo-occipital derecha con leve efecto compresivo sobre la línea media y el cuerno occipital del ventrículo lateral ipsilateral (Figura 1), por lo que se sospechó diagnóstico de síndrome MELAS. Se obtuvieron diferentes trazados espectrales del parénquima sano que se correlacionaron con los resultados de la RMN, lo cual permitió observar una disminución en los picos de N acetil aspartato, colina y creatina, y la aparición de pico dominante de lactato cerebral.
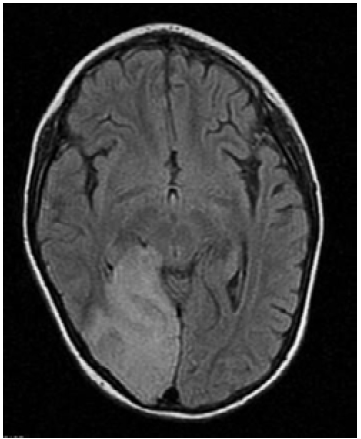
Fuente: Documento obtenido durante la realización del estudio.
Figura 1 Imagen por resonancia magnética cerebral potenciada en T1, corte axial, en la que se observa lesión isquémica a nivel occipital derecho con leve efecto compresivo sobre la línea media y el cuerno occipital del ventrículo lateral del mismo lado.
En los controles clínicos posteriores se evidenció mayor deterioro visual y neurológico; para ese momento la paciente ya era totalmente dependiente de terceros.
Se realizó un estudio del ADNmt con el método AR-MS-qPCR en el Laboratorio de Genética Médica de Baylor College of Medicine, Houston, EE. UU., en el cual se observó la mutación m.3243A>G en el gen MT-TL1. Los niveles de heteroplasmia en las muestras de sangre, orina y epitelio bucal fueron 80%, 97% y 90%, respectivamente. La mutación también se identificó en la madre (37 años) y la hermana (19 años) de la paciente, cuyos niveles de heteroplasmia en sangre, orina y epitelio bucal fueron 16%, 68% y 15%, y 47%, 40% y 57%, respectivamente.
A la paciente se le formuló biotina, carnitina, ubiquinol, creatina, riboflavina, tiamina, vitamina C y vitamina K3; las convulsiones se manejaron con oxcarbazepina, levetiracetam y clobazam. A pesar del tratamiento instaurado, la niña tuvo un rápido deterioro, presentando una involución de las capacidades adquiridas, y falleció a los cuatro años del inicio de los signos clínicos.
Discusión
Las enfermedades mitocondriales son desórdenes neurológicos hereditarios, multisistémicos y progresivos causados por mutaciones en el ADNmt o el ADN nuclear. Estas enfermedades tienen una prevalencia de 16.5 por cada 100 000 habitantes;4 pueden presentar diferentes grados de severidad y manifestaciones clínicas, y el síndrome MELAS es una de las más frecuentes.
Aunque las características clínicas del síndrome MELAS pueden tomar años en aparecer, de manera gradual se presentan alteraciones en el desarrollo cognitivo y motor. Esta enfermedad puede confundirse con encefalitis herpética, ya que frecuentemente debuta con cefalea, confusión mental y fiebre.5 En el 62% de los pacientes la enfermedad comienza antes de los 15 años y en el 75%, antes de los 20 años,3,6,7 por lo que llama la atención la aparición temprana en la paciente reportada.
La causa de los accidentes cerebrovasculares (ACV) en el síndrome MELAS aún no está definida, pues pueden ser ocasionados por citopatías o angiopatías mitocondriales, así como por mecanismos neurovasculares celulares no isquémicos;8 de igual forma, se cree que la hiperexcitabilidad neuronal aumenta las demandas energéticas y genera un desbalance entre la oferta y la demanda de trifosfato de adenosina (ATP) debido a defectos de la fosforilación en neuronas susceptibles, lo que ocasiona necrosis cortical.1 Asimismo, se han observado alteraciones en la cadena transportadora de electrones de la membrana mitocondrial interna, encontrando que al menos el 42%, el 29% y el 23% de los pacientes muestran una disminución de la actividad de los complejos I, III y IV de la cadena respiratoria, respectivamente.2 En los episodios de apoplejía el óxido nítrico se ha encontrado disminuido, ya sea por una producción defectuosa o por secuestro posproducción.7 Los eventos neurológicos en jóvenes son atípicos y a menudo desencadenados por fiebre, deshidratación o estrés; estos pueden incluir cefaleas de tipo migraña o convulsiones.9
Clínicamente, el síndrome MELAS se caracteriza por episodios recurrentes similares a un infarto cerebral isquémico con hemiparesia u otros signos neurológicos focales, y al menos dos de las siguientes condiciones: convulsiones focales o generalizadas, demencia, cefalea migrañosa recurrente y vómito.1,3,10
En las neuroimágenes de pacientes con síndrome MELAS se pueden encontrar lesiones cerebrales similares a un infarto cerebral isquémico -especialmente en las regiones posteriores de los lóbulos temporales, parietales y occipitales- pero que superan un territorio vascular; de igual forma, son evidentes las calcificaciones cerebrales en ganglios basales y la atrofia cerebral desproporcionada para la edad del paciente,9 tal como se corroboró en el caso descrito (Figura 2).
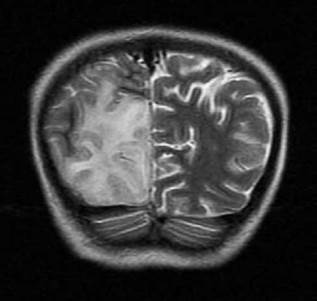
Fuente: Documento obtenido durante la realización del estudio.
Figura 2 Imagen por resonancia magnética cerebral potenciada en T2, corte coronal, en la que se observa lesión isquémica occipital derecha que no se corresponde con ningún territorio vascular.
En pacientes con síndrome MELAS también se ha encontrado que la recaptación de glucosa en el cerebro está disminuida, especialmente en los lóbulos temporal y occipital, y puede o no presentar sintomatología neurológica.11 La hipoperfusión cerebral puede afectar la parte posterior de la corteza del cíngulo cerebral, lo cual es un hallazgo común en la enfermedad de Alzheimer y puede estar relacionado con la demencia que se ha evidenciado en los estados avanzados de esta enfermedad.9
La resonancia magnética por espectroscopia permite detectar áreas focales de acidosis láctica en el cerebro de los pacientes con síndrome MELAS, tal como se evidencia en el presente caso; con los angiogramas cerebrales es posible confirmar la ausencia de alteraciones en los grandes vasos.9 Otras características de esta enfermedad son la hiperalaninemia y el aumento del ácido láctico en sangre y en el líquido cefalorraquídeo.12,13 La relación lactato/piruvato en este tipo de pacientes es mayor al rango de 15 a 25 dado el aumento de la relación nicotinamida adenina dinucleotido hidruro / nicotinamida adenina dinucleótido (NADH/NAD), tanto en la mitocondria como en el citoplasma. El caso reportado muestra un aumento persistente del lactato sérico con alteración de la relación lactato/piruvato no asociado al ejercicio o la ingesta.
En las biopsias musculares de pacientes con síndrome MELAS se espera encontrar fibras rojas rasgadas intercaladas con zonas normales y con hiperactividad para la enzima citocromo C oxidasa, pero hay que tener en cuenta que no todos los casos se acompañan de miopatía y que la mayoría de pacientes pediátricos no muestran alteraciones histológicas, lo que no descarta el diagnóstico.
Por su parte, el electroencefalograma puede mostrar enlentecimiento de la actividad de fondo, de las respuestas fotoparoxísticas y de las descargas periódicas lateralizadas varias semanas después de la instauración del cuadro clínico.1
El síndrome MELAS, por ser una enfermedad mitocondrial, es trasmitido por herencia materna. Esta es una patología en la que se presenta poliplasmia, heteroplasmia, segregación mitótica y efecto umbral con influencia de factores genéticos y ambientales, lo que explica la variabilidad de su presentación. El fenotipo depende de la naturaleza y el porcentaje de ADNmt mutado, ya que si sobrepasan un umbral, la función del sistema de fosforilación oxidativa se compromete y la síntesis de ATP disminuye, presentándose así la enfermedad. Los tejidos que tienen mayor requerimiento energético (cerebral, músculoesquelético y cardiaco) son los más afectados,14 pero el porcentaje de mutación suele ser más bajo en la sangre que en el músculo, siendo este último el tejido preferido para su estudio.1,15
En las enfermedades mitocondriales el porcentaje de mutación puede variar a lo largo de la vida, por lo que las manifestaciones clínicas ocurren en diferentes momentos y afectan diferentes órganos y tejidos.16 Como se mencionó antes, en el síndrome MELAS la mutación más frecuente es la A3243G del gen MT-TL1 en el AD-Nmt que codifica para el ARNt, la cual se encuentra en cerca del 80% de los casos1,2,15, siendo identificada en el caso aquí reportado. Además, como esta es una mutación heteroplásmica, cuanto mayor es el porcentaje de la mutación, más precoz es el inicio de la enfermedad y más grave es su curso; de esta forma, una mutación en el ADNmt del 92% desencadenará el desarrollo de síndrome de Leigh, mientras que una mutación de entre el 74% y el 87% desencadenará el desarrollo de síndrome MELAS.10
Una de las principales manifestaciones del síndrome MELAS es la hemianopsia por ceguera cortical, pero también pueden presentarse crisis epilépticas parciales o generalizadas; episodios recurrentes y transitorios de apoplejía caracterizados por hemiplejía,6,15 y ataxia cerebelosa que puede preceder a la aparición de apoplejía.13 Otras manifestaciones incluyen sordera neurosensorial, neuropatía periférica, síntomas extrapiramidales, disfunción autonómica, confusión, demencia, retraso en el crecimiento, fiebre, intolerancia al ejercicio, miopatía, oftalmoplejía, retinosis pigmentaria, cardiomiopatía hipertrófica o dilatada, defectos en el sistema de conducción cardiaco, diabetes mellitus no insulino-de-pendiente, episodios de pseudoobstrucción intestinal, pancreatitis, disfunción del túbulo renal proximal, síndrome nefrótico, déficit de hormona de crecimiento e hipotiroidismo.1,3,6,8,10
El manejo de este síndrome es multidisciplinario, de soporte y sintomático; además, es un tratamiento encaminado, por un lado, a optimizar las condiciones nutricionales y el estilo de vida de los pacientes, y, por el otro, a evitar o corregir las descompensaciones metabólicas agudas. Las estrategias implementadas buscan disminuir los efectos deletéreos de la función de las cadenas respiratorias anormales, reducir la presencia de agentes tóxicos y corregir la deficiencia de cofactores esenciales, y aunque en la actualidad no hay un consenso específico de tratamiento, algunos ensayos terapéuticos han reportado ciertos beneficios con el uso de corticosteroides, coenzima Q, nicotinamida, riboflavina, L-arginina, citrulina y resveratrol.2,3,7,17
Las vitaminas del complejo B se han considerado como la primera línea de tratamiento en el síndrome MELAS. La riboflavina puede inhibir la ruptura proteica del complejo I de la cadena respiratoria en la membrana interna mitocondrial, y de esta forma incrementar la actividad enzimática y actuar como antioxidante, sin ninguna contraindicación conocida hasta la fecha, aunque puede causar anorexia, vómito y cambios en el olor y color de la orina. Por su parte, la coenzima Q10 ha sido aprobada por la Food and Drug Administration para el tratamiento de esta enfermedad con la indicación de suministrarla en forma de ubiquinol, en dosis ascendentes e idealmente con la comida. Aunque no se han confirmado beneficios y aún son pocos los datos científicos que respaldan su uso, la tiamina, las vitaminas C y E y la carnitina también han sido usadas empíricamente; esta última en combinación con otras enzimas y cofactores.3,17
El pronóstico del síndrome MELAS es generalmente pobre, especialmente cuando el inicio es temprano y con compromiso multiorgánico;3,10 además, esta es una enfermedad con alta morbimortalidad, relacionada especialmente con encefalopatía, cardiomiopatía y falla renal.18
Conclusiones
Los errores innatos del metabolismo, y dentro de ellos el síndrome MELAS, comprenden un conjunto amplio de enfermedades que de manera aislada pueden ser infrecuentes, pero que en su totalidad registran un alto número de casos. Su presentación varía dependiendo de la vía metabólica afectada y muchos de ellos tienen diferentes formas de presentación y manifestaciones progresivas. En este sentido, las enfermedades mitocondriales pueden confundirse con otras patologías mucho más frecuentes, por lo que deben ser consideradas en pacientes con antecedentes de epilepsia y otras alteraciones neurológicas como ataxia e involución del neurodesarrollo. La confirmación de su diagnóstico, pues amerita consejería genética.

