











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
INTRODUCTION
The poultry protein industry in Brazil is of significant global importance, ranking as the second-largest chicken meat producer in the world. Recent data reveals that the annual production of chicken meat has reached 14.524 million tons, which is exported to 151 different nations, resulting in an export value of $7.6 billion (ABPA, 2022).
Due to the high demand in this sector, the poultry industry relies on raising large numbers of birds. In this context, it is also necessary to adopt effective alternatives for disease prevention and control and improve the growth performance of poultry. Bacterial infections are among the most common diseases in poultry farming, capable of impacting animal health and product quality, in addition to posing a risk to public health. Examples of bacterial poultry diseases include fowl typhoid (Salmonella gallinarum), colibacillosis (Escherichia coli), pasteurellosis (Pasteurella multocida), infectious coryza (Avibacterium paragallinarum), and others (EMBRAPA, 2021). These diseases also raise substantial concerns as they have the potential to cause significant economic losses, affecting both producers and the economy as a whole (Gutarowska et al., 2018; Paudel et al., 2021; Gržinić et al., 2023).
Strategies aimed at reducing the incidence of diseases include the prophylactic use of antimicrobials in humans and animals to combat bacterial infections. Additionally, these agents are administered sub-therapeutically as feed additives, aiming to promote animal growth. Throughout the twentieth century, the use of antibiotics was a significant factor in reducing mortality rates associated with infectious diseases. However, the indiscriminate and inappropriate use of these drugs emerged as one of the main underlying causes of the emergence of multidrug-resistant (MDR) bacteria (Moyane et al., 2013; Baynes et al., 2016; Manyi-Loh et al., 2018; Berghiche et al., 2018; Arbab et al., 2021; Moretto et al., 2021).
The molecular profile and antimicrobial resistance of bacterial isolates are essential for understanding the genetic diversity and resistance of microorganisms (Arbab et al., 2021; Deusdará et al., 2023). Molecular identification involves techniques such as PCR and whole-genome sequencing, which are based on DNA amplification. PCR is used to amplify specific regions of each species, employing different primers, including genes encoding the 16S rRNA and 23S rRNA subunits (Zarrilli et al., 2013). This approach is crucial for identifying pathogens, assessing treatment efficacy, and implementing preventive measures (Gomes et al., 2019; Arbab et al., 2021). Additionally, it helps reduce the risk of the spread of multidrug-resistant pathogens, benefiting animal health, food safety, and public health (Fardsanei et al., 2017; Moretto et al., 2021).
Given the challenges associated with poultry farming, producers need to develop and implement comprehensive prevention plans. This study aims to identify pathogenic microorganisms and analyze the antibiotic susceptibility of strains isolated from poultry environments in the southern region of Tocantins, Brazil. Consequently, this approach makes a significant contribution to promoting animal health and developing appropriate public health strategies. Furthermore, the study fosters knowledge dissemination, as data on the evaluation of antibiotic-resistant bacteria in poultry farms in southern Tocantins is scarce in the literature.
MATERIALS AND METHODS
Sample Collection and Processing. The samples were collected from chicken poultry farms of the species Gallus gallus domesticus in rural property Tocantins, Brazil (11°47'48'' S and 49°31'44''W). These were identified with sequential codes of poultry farm isolates: IGA1, IGA2, IGA3, IGA4, and IGA5. These were obtained using sterile microbiological swabs at different locations, such as soil, chicken coop floor, and fecal samples, which were subsequently stored in sterile containers and transported to the laboratory. Sample processing was carried out immediately, and the samples were cultured in a Brain Heart Infusion (BHI) culture medium at 37°C for 24 hours in a Biochemical Oxygen Demand (BOD) incubator. After incubation, the resulting colonies were identified using the 16S RNA gene and the analysis of the antibiotic susceptibility profile.
DNA Extraction, Amplification, and Sequencing. Genomic DNA extraction from bacterial isolates was performed using a commercial kit (Promega, Madison, WI, USA) and silica columns. In summary, DNA was extracted from the collected isolates and added to sterile tubes. The biological material was centrifuged at 14,000 rpm for 1 minute, and the supernatant was removed. The material was homogenized in a lysis solution with extraction buffer and proteinase K, incubated at 55 °C for 30 minutes, and centrifuged at 12,000 rpm for 3 minutes. The supernatant was transferred to a new tube containing isopropanol, and after centrifugation at 12,000 rpm for 2 minutes, the precipitated DNA was collected. The quantity and quality of the extracted DNA were assessed by agarose gel electrophoresis (1%) with a molecular weight marker and intercalating dye. DNA purification involved cell lysis, DNA binding to the silica columns of the kit, washing to remove impurities, and elution of the purified DNA in the elution buffer. The obtained DNA was stored at -20 °C for later analysis.
Amplification of the 16S rRNA gene was carried out using specific primers that recognize conserved sequences at the ends of the gene. The reaction mix contained 10µL of reaction buffer, 2µL of universal primers 27F (5'-AGAGTTTGATCMTGGCTCAG-3') and 1492R (5'-GGTTACCTTGTTACGACTT-3') (10µM each), 1µL of dNTPs (10 mM), 0.5µL of Taq DNA polymerase (5 U/µL), and 1µL of genomic DNA. The mix was pipetted into PCR tubes and placed in a thermal cycler for amplification.
The amplified DNA was subjected to a sequencing reaction using fluorescent chain terminators (Sanger et al., 1977). The sequencing reaction was prepared with 10µL of amplified DNA sample, 4µL of sequencing buffer, 2µL of sequencing primer (10µM), and 4µL of BigDye Terminator v3.1 kit from Thermo Fisher Scientific. The mix underwent a thermal cycle: 1 minute at 96°C, followed by 25 cycles of 10 seconds at 96°C, 5 seconds at 50°C, and 4 minutes at 60°C. After sequencing, the mixture was purified using the BigDye XTerminator kit (Thermo Fisher Scientific), precipitated with ethanol, washed with 70% ethanol solution, resuspended in ultrapure water, and subjected to capillary electrophoresis in the Applied Biosystems 3730XL sequencer (Thermo Fisher Scientific). The obtained sequences were analyzed with Sequencing Analysis v5.4 software, with quality assessment using Phred software. Low-quality sequences (Phred < 20) were removed from the analysis, and the remaining sequences were aligned and assembled into contigs using CodonCode Aligner v.11.0.3 software.
Characterization of the antibiotic susceptibility profile. The disk diffusion method was employed to evaluate bacterial susceptibility and resistance classification to antibiotics, following the guidelines of the Clinical and Laboratory Standards Institute (CLSI, 2020). The isolates were cultured in 100 mL of TSB broth (Soybean Tryptone Broth) under agitation at 120 rpm at 37°C for 8 hours. Subsequently, the growth was adjusted to 108 CFU/mL and seeded on Mueller-Hinton agar (Kasvi, Italy). A disk of antibiotics was then inoculated onto the plate and incubated at 37°C for 18 to 24 hours to record the formation of the inhibition zone. The antibiotics (Sensifar®-Brazil) tested were: ampicillin/sulbactam (20µg), amikacin (30µg), ceftazidime (30µg), cefepime (30µg), ciprofloxacin (5µg), cotrimoxazole (25µg), gentamicin (10µg), imipenem (10µg), levofloxacin (5µg), linezolid (30µg), meropenem (10µg), minocycline (30µg), norfloxacin (10µg), piperacillin/tazobactam (110µg), polymyxin B (330 IU), tetracycline (30µg), vancomycin (30µg), These antibiotics allow for a broader assessment of pathogen resistance, providing a better understanding of the effectiveness of treatments. The inhibition zone was measured manually using a metric ruler across the zone inhibition around each antibiotic and classified in resistance to ≥ 1 antimicrobial agent and in ≥ 3 antimicrobial categories considered MDR (Multidrug Resistance) (Magiorakos et al., 2012; Deusdará et al., 2023).
Bioinformatics analysis. The DNA sequences are imported into CodonCode Aligner V4.2.7 software (LI-COR®) and undergo a series of steps, including the identification of overlapping regions, the removal of low-quality regions, and the clustering of similar sequences to produce a consensus sequence. To identify and compare DNA sequences in the NCBI and RDP databases, sequences were first uploaded in FASTA or GenBank format using search tools such as BLAST (Basic Local Alignment Search Tool) and the Ribosomal Database Project (RDP) (Altschul et al., 1990; Cole et al., 2014).
The construction of the phylogenetic tree by dendrogram was performed using MEGA 11 software using multiple alignments of DNA sequences with the MUSCLE algorithm and the analysis of evolution models with the Bayesian information criterion (BIC). The phylogenetic tree was constructed using the Neighbor-Joining (NJ) method using the distance matrix corrected by the chosen model, and finally, the robustness of the tree was assessed through bootstrap analysis with 1000 replicates. The resulting tree was visualized and edited in FigTree software (Kumar et al., 2018).
RESULTS AND DISCUSSION
Bacterial identification using 16S rRNA. A total of five isolates were identified from biological samples (feces) and environmental samples (floor and perch) of poultry farms (Table 1). Isolates IGA1, IGA2, IGA3, IGA4, and IGA5 were identified through the sequencing of the 16S rRNA gene and were found to belong to different bacterial genera (Table 1). In the phylogenetic analysis, isolate IGA1 exhibited 18 characteristic nucleotide sequences of the species G. creatinolyticus (Table 1; Figure 1). A total of 1.408 positions in the final dataset of isolate IGA1 showed 99.4% similarity to G. creatinolyticus in BLAST, clustering in the clade with 100% bootstrap support (Figure 1). The isolate IGA2 presented 15 characteristic nucleotide sequences (Table 1; Figure 2). In the final dataset, 1.437 positions of the isolate showed 99.86% similarity to E. gallinarum based on BLAST analysis, clustering within a clade that was supported by 88% bootstrap (Figure 2). IGA3 exhibited 18 distinctive nucleotide sequences characteristic of the genus Enterobacter sp. There were a total of 1414 positions in the final dataset of isolate IGA3 that showed 99.5% similarity to Enterobacter mori species in BLAST, clustering in the clade with 86% bootstrap support (Table 1; Figure 3). IGA4 displayed 18 characteristic nucleotide sequences of the species L. fusiformis (Table 1; Figure 4). There were a total of 1422 positions in the final dataset of isolate IGA4, demostrated 99.72% similarity to the bacterium L. fusiformis in BLAST, clustering in the clade with 91% bootstrap support (Figure 4). IGA5 showed 21 characteristic nucleotide sequences of the genus and species E. faecalis. The final dataset, comprising 1.427 positions, showed 100% similarity (Table 1; Figure 5).
Antibiotic susceptibility testing. The susceptibility and resistance profiles of the five isolates revealed varied patterns for the different antimicrobial agents tested. Isolate IGA1 demonstrated resistance to the antimicrobials imipenem (IPM10), meropenem (MER10), ceftazidime (CAZ30), trimethoprim (TRI5), and doxycycline (DOX30) from the cephalosporin, carbapenem, sulfonamide, and tetracycline classes, thus being classified as MDR (multidrug-resistant) (Table 1). IGA2 showed resistance to different antimicrobial agents tested, including ceftazidime (CAZ30), cefepime (CPM30), doxycycline (DOX30), imipenem (IPM10), meropenem (IPM10), piperacillin-tazobactam (PPT110), and tetracycline (TET30), which belong to the cephalosporin, carbapenem, penicillin, and tetracycline classes (Table 1). However, it is important to note that isolate IGA2 exhibited resistance to β-lactam antibiotics (cephalosporin, carbapenem, and penicillin). IGA3 showed resistance to the antimicrobials aztreonam (ATM30) and trimethoprim (TRI5), while no resistance was observed for the others (Table 1). Isolate IGA4 demonstrated resistance to only two antimicrobials, ceftazidime (CAZ30) and cefepime (CPM30), from the cephalosporin class, and sensitivity to most antimicrobials, including those belonging to the fluoroquinolone class such as ciprofloxacin (CIP5) and norfloxacin (NOR10). Additionally, it showed sensitivity to linezolid (LNZ30) and gentamicin (GEN10). On the other hand, strain IGA5 did not exhibit resistance to any tested antimicrobials (Table 1).
Table 1 Identification of microorganisms by 16S rRNA sequencing compared to GenBank NCBI and antimicrobial resistance.
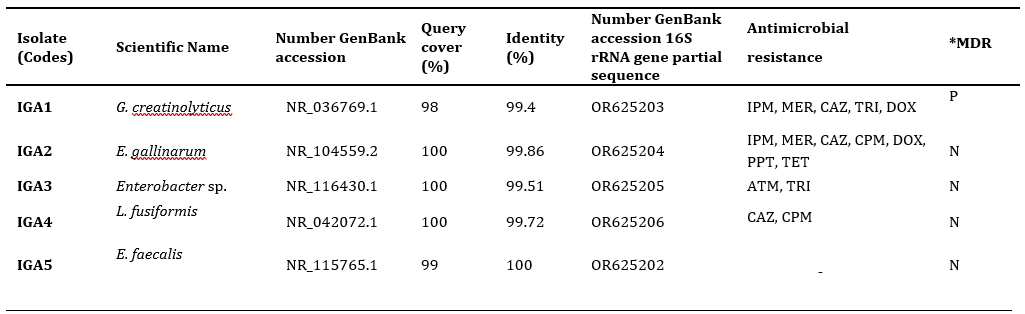
*Antibiotics: imipenem (IPM), meropenem (MER); ceftazidime (CAZ), cefepime (CPM), trimethoprim (TRI), doxycycline (DOX), aztreonam (ATM), piperacillin/tazobactam (PPT), tetracycline (TET). *Multidrug-resistant bacteria. P: Positive; N: Negative.

Figure 1 Phylogenetic trees obtained by the neighbor-joining method with the Jukes-Cantor parameter and Bootstrap of 1000 replicas of 16S rRNA gene sequences of the isolate 2023.0290.16S (IGA1) with GenBank accession number OR625203.

Figure 2 Phylogenetic trees obtained by the neighbor-joining method with the Jukes-Cantor parameter and Bootstrap of 1000 replicas of 16S rRNA gene sequences of the isolate 2023.0291.16S (IGA2) with GenBank accession number OR625204.

Figure 3 Phylogenetic trees obtained by the neighbor-joining method with the Jukes-Cantor parameter and Bootstrap of 1000 replicas of 16S rRNA gene sequences of the isolate 2023.0292.16S (IGA3) with GenBank accession number OR625205.

Figure 4 Phylogenetic trees obtained by the neighbor-joining method with the Jukes-Cantor parameter and Bootstrap of 1000 replicas of 16S rRNA gene sequences of isolate 2023.0293.16S (IGA4) with GenBank accession number OR625206.

Figure 5 Phylogenetic trees obtained by the neighbor-joining method with the Jukes-Cantor parameter and Bootstrap of 1000 replicas of 16S rRNA gene sequences of isolate 2023.0289.16S (IGA4) with GenBank accession number OR625206.
Bacterial infections can significantly impair animal health and performance, leading to serious complications, especially when there is resistance to multiple antimicrobials (MDR), which complicates treatment and increases the risks of more severe outbreaks. Therefore, evaluating the resistance profile of bacteria in poultry farms is important since they can be sources of pathogenic microorganisms (Lateef et al., 2015; Deusdará et al., 2023). Thus, using the 16S rRNA gene sequencing, it is possible to evaluate the presence of species of prokaryotic organisms, such as bacteria, fungi, and archaea, and its ability to provide valuable phylogenetic and taxonomic information about microbial diversity, a relevant tool for identifying pathogens in poultry farms (Deusdará et al., 2023).
In the scope of this study, these approaches were implemented, allowing for the characterization of antibiotic resistance. Furthermore, accurate identification of the investigated microorganisms validates the reliability of the 16S rRNA gene sequencing technique for constructing the phylogenetic profile and determining the isolates G. creatinolyticus (IGA1), E. gallinarum (IGA2), Enterobacter sp. (IGA3), L. fusiformis (IGA4), and E. faecalis (IGA5). G. creatinolyticus (IGA1) exhibited multidrug-resistant phenotypes to antibiotics. Is considered ubiquitous, as it can be found in various habitats, including different types of soil, clinical samples, cheeses, and plants, and plays an important role in various biotechnological processes. However, when pathogenic, it is associated with urinary tract infections and bacteremia. This species may be related to opportunistic infections in humans and animals, especially in immunocompromised individuals, and its presence in infections may have significant clinical implications (Hou et al., 1998; Santos et al., 2020). In the case of E. gallinarum (IGA2), it is becoming increasingly relevant as pathogens, both in hospital settings and in community-acquired infections. Usually considered of low pathogenic potential, these species are recognized as opportunistic pathogens in humans, particularly in immunocompromised individuals, and in animals, with the ability to cause severe invasive infections such as endocarditis, bacteremia, urinary tract infections, and pelvic infections (Ruoff et al., 1990; Reid et al., 2001; Monticelli et al., 2018).
Enterobacter sp. (IGA3) showed resistance to antibiotics from the monobactam and sulfonamide classes. The Enterobacter genus, belonging to the Enterobacteriaceae family, is primarily associated with healthcare-associated infections. Currently, there are 22 species of Enterobacter, although not all are recognized to cause diseases only in humans. These species have played a significant role in the origin of various infections, such as urinary tract infections (UTIs), respiratory infections, soft tissue infections, osteomyelitis, and endocarditis, along with other clinical manifestations. These infections predominantly occur in healthcare settings, although they can also be acquired in the community (Wilberger et al., 2012; Davin-Regli & Pagès, 2015).
On the other hand, L. fusiformis (IGA4) exhibited resistance to two antibiotics, ceftazidime and cefepime, from the cephalosporin class. This bacterial genus can be found in soil, water, and clinical environments, including hospitals. Although it is an invasive bacterium, it is rarely associated with diseases in humans. However, isolated cases of severe sepsis due to persistent bacteremia caused by Lysinibacillus have been reported (Wenzler et al., 2015). And, finally, E. faecalis (IGA5) showed no resistance to the evaluated antimicrobials. However, the relevance of this species as an opportunistic pathogen in birds and humans is highlighted, emphasizing the continued importance of monitoring and controlling antimicrobial resistance associated with E. faecalis (Pereira et al., 2020). And some strains have been used as animal probiotics.
Research like this should be more extensively explored by other researchers, given the limited availability of data in the literature that contribute to the identification of bacteria in poultry farms. Moreover, these studies provide valuable information on management practices and appropriate treatments to ensure the health and welfare of birds.
CONCLUSIONS
This study highlighted the importance of identifying the molecular profile through the application of the 16S rRNA molecular profiling technique and evaluating antimicrobial sensitivity in bacterial isolates from poultry farms. Identifying bacteria with pathogenic potential in poultry facilities can provide crucial information for monitoring and disease control in birds. Further research is needed to perform comprehensive genotyping of bacterial isolates, including virulence and resistance gene analyses. These data play a fundamental role in prevention strategies, aiming to improve poultry health, ensure food safety, and protect both animal and public health.

