











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
Los begomovirus (familia Geminiviridae) poseen un genoma constituido por ADN cadena sencilla circular cuyo tamaño oscila entre 2,5 y 2,7 kb. Los virus de género Begomovirus son mayoritariamente bipartitas, es decir, poseen dos componentes genómicos constituidos por moléculas de ADN denominadas AyB, las cuáles se encapsidan separadamente. Son transmitidos por mosca blanca Bemisia tabaci y afectan plantas dicotiledóneas (Brown et al., 2015). A la fecha hay reportadas 409 especies de virus pertenecientes a este género geminiviral, el cual se considera el más abundante que afecta plantas, superando ampliamente a los virus RNA pertenecientes al género Potyvirus (ICTV, 2018). Hoy por hoy son considerados una de las principales limitantes en la productividad agrícola que ocasiona reducción en la producción o pérdidas totales de las cosechas (Hernández-Espinal et al., 2018). Tal es el caso de los virus rizado amarillo de la hoja del tomate (tomato yellow leaf curl virus, TYLCV) y del mosaico de la yuca africana (African cassava mosaic virus, ACMV), quienes tienen un gran impacto en la agricultura por las cuantiosas pérdidas económicas generadas en cultivos de tomate y yuca, respectivamente (Scholthof et al., 2011); lo que ha despertado el ferviente interés científico para ser caracterizados a nivel molecular (Fondong, 2013)
Gracias a mecanismos evolutivos como la recombinación o pseudo-recombinación los begomovirus han emergido e incrementado su diversidad, hecho que ha acentuado su importancia patogénica en las zonas tropicales y subtropicales del mundo particularmente en las familias botánicas Fabaceae, Euphorbiacea, Solanaceae y Malvaceae (Brown y Bird, 1992; Jones, 2003). Este incremento en diversidad y patogenicidad que se ha verificado en los begomovirus ha sido por el papel que como hospedero alterno de estos virus han jugado las plantas arvenses, particularmente, durante la rotación de los cultivos (Ambrozevicius et al., 2002; Jovel et al., 2004; Sobrinho et al., 2014). Reforzando los argumentos de las arvenses como fuente de inóculo de virus para favorecer su persistencia y propagación (Duffus, 1971; Marwal et al., 2014); así como escenarios naturales en donde fenómenos de recombinación genética podrían verificarse favoreciendo la generación de nuevas variantes virales con mayor patogenicidad y adaptación a nuevos hospederos en un corto lapso de tiempo (Padidam et al., 1999; Da Silva et al., 2011). Un buen ejemplo de ello se registra en Brasil, con el virus del mosaico dorado del tomate (tomatogolden mosaic virus, TGMV), cuyas recombinaciones y pseudo-recombinaciones con otros begomovirus presentes en plantas arvenses asociadas al cultivo de tomate ha conducido a un aumento en la misma diversidad del TGMV, a tal punto que hoy en día bien se puede hablar del complejo del virus del mosaico dorado del tomate (Faria et al., 1997; Ribeiro et al., 1998; Inoue-Nagata et al., 2016)
En el departamento del Valle del Cauca (Colombia) se ha detectado la presencia begomoviral en las siguientes arvenses asociadas al cultivo de tomate: Momordica charantia (Cucurbitaceae); R. humilis y Petiveria alliaceae (Phytolaccaceae); Laportea estuans (Urticaceae); A. dubius (Amaranthaceae); Lantana cámara y Verbena sp. (Verbenaceae); Croton hirtus (Euphorbiaceae); Hybanthus attenuatus (Violaceae); Plumbago sp. (Plumbaginaceae); y Caesalpinia sp., Desmodium sp. y R. minima (Leguminosae) (Vaca-Vaca et al., 2011; López-López et al., 2012; López-López et al., 2014). Sin embargo, a la fecha sólo ha sido reportado la obtención del genoma completo de un begomovirus aislado de C. hirtus, el cuál fue denominado virus del mosaico dorado de Croton (croton golden mosaic virus, CroGMV) (Vaca-Vaca et al., 2018). Con base en lo anterior se hace necesario establecer las identidades moleculares plenas de otros begomovirus previamente detectados en las otras arvenses. La comprensión de la diversidad begomoviral presente en las arvenses que acompañan cultivos de interés económico, como lo es el tomate, permitirá a futuro no solo construir la virosfera de este género viral, sino además tomando como base este conocimiento establecer programas de mejoramiento para esta solanácea en procura de la obtención de resistencia a estos nuevos begomovirus que algún día podrían emerger desde las arvenses, constituyéndose en una limitante para la propia producción del tomate.
Por tanto, el objetivo de esta investigación fue obtener el genoma completo de un begomovirus bipartita presente en cinco arvenses A. dubius, R. humilis, R. minima, Desmodium sp. y Caesalpinia sp., las cuales fueron colectadas en los alrededores de cultivos de tomate presentes en los municipios de Ginebra y Cerrito en el departamento del Valle del Cauca.
MATERIALES Y MÉTODOS
Colección de muestras
Investigaciones previas realizadas por nuestro grupo de investigación, IPMA Interacción Planta Microorganismo Ambiente, identificaron como positivas para begomovirus a las arvenses R. minima (muestras AT35 y AT36), R. humilis (AT38), Desmodium sp. (AT40) y Caesalpinia sp. (AT41) que fueron colectadas en cultivos de tomate en el municipio de Cerrito; y a A. dubius (AT32) que fue colectada en el municipio Ginebra, Valle del Cauca (López-López et al., 2014) (Fig. 1).

Extracción de ADN genómico
Se partió de aproximadamente 1 cm2 de material vegetal de tejido vegetal foliar seco para llevar a cabo la extracción de ADN total empleando para ello el kit de extracción Dneasy Plant Mini Kit (Qiagen®), siguiendo las recomendaciones del fabricante.
Detección de begomovirus en las arvenses por PCR
Para determinar la presencia de begomovirus en las muestras colectas en campo se utilizaron los oligos PAR1c496/PAL1v1978 y PBL1v2040/PCRc1 (Rojas et al., 1993) que amplifican un fragmento de aproximadamente ~ 1,1 kb y 0,5 kb correspondientes a los componentes genómicos A y B, respectivamente. Las condiciones del PCR para ambos pares de oligos se establecieron teniendo en cuenta lo reportado por Rojas et al., (1993) y se utilizó un termociclador iCycler (BioRad®). Como control positivo se empleó un fragmento del virus del mosaico amarillo de la papa (PYMV), un begomovirus endémico de Colombia que limita la producción de esta solanácea a lo ancho y largo del país (Vaca-Vaca et al., 2012). Los fragmentos amplificados del ADN viral fueron sometidos a electroforesis en geles de agarosa al 0,8 % y posteriormente se visualizaron en un transiluminador (Molecular Imager Gel DocXR+ Systems BIORAD®) utilizando el software Quantity-One® 4.6.5 provisto por el fabricante del equipo.
Amplificación y clonación del genoma completo A y B del begomovirus
Se amplificó el título viral presente en las arvenses utilizando el kit TempliPhi™ 100 Amplification (GE Healthcare®), en una reacción isotérmica de 30 °C durante 22 horas siguiendo las condiciones propuestas por Inoue-Nagata et al., (2004). Este kit contiene la ADN polimerasa del bacteriófago Φ 29 que permite amplificar genomas circulares de begomovirus, empleando la técnica de amplificación por circulo rodante (RCA). Se confirmó la presencia de los genomas virales amplificados corriendo un gel de agarosa 1 %. Posteriormente, los productos amplificados fueron cortados diferentes enzimas de restricción para linealizar el genoma geminiviral A y B, respectivamente, a 2,7 kb aproximadamente. A continuación, los fragmentos de 2,7 kb fueron ligados con la enzima T4 DNA ligasa (Invitrogen®) al plásmido pBluescript KS+ (Stratagene®) previamente desfosforilado con la enzima FastAP (ThermoFisher®).
Las moléculas recombinantes se transformaron en células competentes de Escherichia coli (One Shot, Invitrogen®) por el método de choque térmico (Sambrook y Russell, 2001) y posterior selección por antibiótico. La evaluación de la presencia del inserto se realizó por lisis rápida de ADN plasmídico, las cuales arrojaron las posibles colonias que contenían el inserto. A continuación, se extrajo su ADN plasmídico empleado el kit QIAprep Spin Miniprep Kit (Qiagen®) y se realizó una digestión enzimática con la enzima utilizada para clonar, la cual permitió la liberación del inserto clonado.
Los fragmentos clonados fueron enviados a secuenciar en Macrogen Inc. (Seúl, Corea del Sur), empleando la metodología de Sanger. Para terminar de secuenciar el fragmento de 2,7 kb se diseñaron oligos internos empleando la estrategia de "primer walking'.
Análisis de los genomas A y B del begomovirus
Los continuos (contigs) de las lecturas de secuenciación de cada genoma, A y B, fueron generados utilizando el software CLC Main Workbench, v7.5 (Qiagen®). A continuación, se identificaron en cada genoma A y B ensamblado, los marcos de lectura abiertos (Open reading frame, ORF), la secuencia de aminoácidos de cada proteína codificada por cada ORF y la región intergénica (IR).
Identidad viral
Para la clasificación taxonómica del begomovirus aislado se siguió el actual criterio taxonómico de ICTV que establece un porcentaje de identidad <91 % a nivel de nucleótidos del genoma A, el cual demarca una especie de begomovirus nuevo (Brown et al., 2015). Para ello, se seleccionaron las secuencias de nucleótidos de diez begomovirus que presentaron el mayor porcentaje de identidad con el begomovirus aislado en este estudio, empleando el algoritmo BlastN y la base de datos general (nr/nt) del Centro Nacional para Información Biotecnológica (www.ncbi.nlm.nih.gov). Adicionalmente, se seleccionaron begomovirus previamente reportados en Colombia, con el objetivo de realizar un alineamiento múltiple de secuencia de nucleótidos. Las secuencias se editaron en Bioedit y los alineamientos se corrieron Clustal W (Thompson etal., 1997). Finalmente, para determinar los valores de identidad entre estas secuencias se utilizó el software Sequence Demarcation Tool SDT 1.2 (Muhire et al., 2014).
Identificación de elementos cis-regulatorios en la región intergénica (IR).
Se buscaron manualmente los siguientes elementos en la IR de cada genoma A y B: iterones como secuencias repetidas de 8 a 12 nucleótidos ricos en G ubicados generalmente entre el sitio de inicio de la transcripción y antes de la caja TATA de Rep, caja TATA y secuencia de nonanucleótidos, típica de losgeminivirus (Argüello-Astorga et al., 1994). Adicionalmente, se realizó un alineamiento múltiple de secuencias empleando la IR de begomovirus seleccionados del GenBank. Los begomovirus utilizados fueron: Squash leaf curl virus- SQLCVE (M38183), Rhynchosia golden mosaic Yucatan virus- RhGMYV (KP641347), Tomato severe leaf curl virus- ToSLCV (JN676151), Squash mild leaf curl virus- SMLCuV (AF421552), Euphorbia mosaic virus- EuMV (KJ647290), Euphorbia mosaic virus- EuMV (DQ395342), Euphorbia mosaic virus- EuMV (FJ807782), Tomato mildyellowleafcurl Aragua virus-ToMYLCV (AY927277), Euphorbia mosaic Venezuela virus- EuMV (JN368145), Pepper golden mosaic virus- PepGMV (LN848774), Capraria yellow spot Yucatan virus- CarYSYV (KC426927), y Chenopodium leaf curl virus- ChLCV (HM626516).
Relaciones filogenéticas.
La construcción de los arboles filogenéticos se realizó empleando los genomas A y B completos del begomovirus en el programa MEGA 7 (Kumar et al., 2016), empleando el alineamiento múltiple de secuencias de nucleótidos previamente realizado. La reconstrucción filogenética se realizó por Neighbor Joining (Saitou y Nei, 1987) y el patrón de similitud genética fue calculado con el modelo de distancia genética de Tamura-Nei (Nei, 1978).
Diseño de primers específicos para confirmar la presencia del virus RhGMCV en cinco arvenses
Utilizando el software CLC Main Workbench, v7.5 (Qiagen®) se diseñaron un par de oligonucleótidos sobre el genoma A de RhGMCV para amplificar un fragmento de 1,82 kb que porta los genes AR1-AL2-AL3-AL1: AT35F: 5'-AAT-TTG-ATA-AGG-CCG-CTG-3' / AT35R: 5'-GGG-GAA-CAA-AGG-CAA-ATA-AG-3'. Estos oligos fueron utilizados para detectar la presencia de RhGMCV en cada una de las arvenses por PCR. La reacción de PCR se realizó en un volumen de 25 ul que incluía los siguientes componentes: 1,0 μM de cada oligo, 0,2 mM de una mezcla de dNTPs, buffer 1X, 1,25 U Taq polimerasa (Fermentas), 500 ng ADN genómico total de cada arvense y agua Milli-Q hasta completar el volumen final. Se utilizó un termociclador BioRad® (Modelo C1000), con las siguientes condiciones de amplificación: 35 ciclos, de 1 min de desnaturalización a 94 °C, 1 min de alineamiento a 55 °C, y 2 min de extensión a 72 °C. Como control positivo se utilizó ADN genómico de R. mínima (AT35); y como control negativo, agua Milli-Q. Los fragmentos de PCR amplificados fueron visualizados en geles de agarosa al 1,0 %.
RESULTADOS Y DISCUSIÓN
Obtención del genoma completo de un begomovirus presente en cinco arvenses
Previamente nuestro grupo de trabajo detectó y aisló un fragmento viral de 1,2 kb de las arvenses R. minima (AT35 y AT36), R. humilis (AT38), Desmodium sp. (AT40), Caesalpinia sp. (AT41) y A. dubius (AT32) (Fig. 1); y el análisis de la secuencia de nucleótidos de los seis fragmentos begomovirales reveló una identidad entre ellos de 99 %. Con base a estos resultados se seleccionó la arvense R. minima para obtener el genoma completo, ya que presentaba dos muestras positivas y alto título viral durante la detección por PCR (datos no mostrados). A partir del ADN genómico total de R. minima se amplificó el virus por RCA y empleando las enzimas EcoRI y EcoRV se obtuvieron fragmentos de 2,7 kb de los genomas A y B, respectivamente, que fueron clonados. Se obtuvieron tres clonas EcoRI y cinco clonas EcoRV que fueron secuenciados y analizados a nivel de nucleótidos.
El análisis de nucleótidos de los tres fragmentos EcoRI (correspondiente al componente genómico begomoviral A) mostró que eran idénticos entre sí y que presentaban un tamaño de 2584 nt; este componente genómico codificaba para cinco marcos de lectura abiertos (ORFs) y poseía una región intergénica (IR) de 307 nt no codificante en donde se alojaban dos promotores divergentes. En el sentido del virión se identificó un ORF, el gen AV1, que presenta un tamaño de 756 nt y que codifica para una proteína de 251 aa que corresponde a la proteína de la cápside (CP). Mientras que en el sentido opuesto al sentido del virión se identificaron cuatro diferentes ORFs: el primero de estos, tiene un tamaño de 1086 nt y codifica para una proteína de 349 aa, correspondiente a la proteína asociada a la replicación (Rep); a continuación de este se encuentra el segundo ORF tiene un tamaño 390 nt y codifica para una proteína de 132 aa, correspondiente a una proteína con cualidades propias de un transactivador transcripcional (TrAP) de los genes tardíos virales (CP y los genes del movimiento BL1 y AR1 respectivamente); río abajo de TrAP, se encuentra un tercer ORF de 399 nt que codifica para una proteína de 121 aa, correspondiente a una proteína con capacidad de acentuar la replicación viral (REn); por último se encuentra AC4 de 366 nt que codifica una proteína de 121 aa que funciona como determinante de patogenicidad.
El análisis de nucleótidos de los cinco fragmentos EcoRV reveló que eran idénticos entre si y correspondían al componente genético B begomoviral típico. Las cinco clonas tenían un tamaño aproximado de 2543 nt en el que se encontraron una RI de 760 nt, así como dos ORF. En sentido del virón, se identificó el gen BV1 con un tamaño de 882 nt y que codifica para una proteína de 293 aa que participa en el transporte del virión a corta distancia es decir desde núcleo hasta el citoplasma de la célula vegetal del hospedero. Mientras que en el sentido opuesto al sentido del virión, se identificó otro ORF correspondiente al gen BC1 el cual tiene un tamaño de 873 nt y codifica para una proteína de 290 aa, que participa en el movimiento del virión a larga distancia es decir célula a célula.
Caracterización molecular del genoma del begomovirus aislado de R. minima
Para determinar la identidad del begomovirus aislado de R. minima, el componente genómico A completo (2584 nt) se comparó con las secuencias de otros begomovirus reportados en Colombia y con aquellos con mayor identidad depositadas en bases de datos (> 81 %). En la Tabla 1 se observan los valores obtenidos del alineamiento múltiple con el software SDT para el componente genómico A, el cuál presentó la mayor identidad (90,9 %) con el virus del mosaico dorado de Rhynchosia de Yucatán (RhGMYV, KP641347) y un 89,1 % con el virus del mosaico de Rhynchosia de Sinaloa (RhMSV, DQ406672), aislados de la arvense R. minima en Yucatán y Sinaloa, respectivamente, localizados en México. Tomando como base el criterio de demarcación actual para las especies de Begomovirus establecido por el Comité Internacional de Taxonomía de Virus, ICTV, en donde claramente se establece que una identidad inferior al 91 % a nivel del componente genómico A completo es considerada una especie nueva (Brown et al., 2015), podemos concluir que el begomovirus aislado de R. minima en Valle del Cauca (Colombia) es una especie begomoviral nueva. Con base en este resultado se propone el nombre de Virus del mosaico dorado de Rhynchosia de Colombia (Rhynchosia golden mosaic Colombia virus, RhGMCV) para designar a esta nueva especie begomoviral, teniendo en cuenta: los síntomas observados en campo, la arvense de donde se colectó por primera vez, R. minima así como la localidad donde ésta se encontró. La secuencia de nucleótidos del genoma de Virus del mosaico dorado de Rhynchosia de Colombia (RhGMCV) fue depositada en la base de datos GenBank, donde se puede identificar con el número de accesión KX216480, correspondiente al componente genómico Ayel número KX216481, correspondiente al componente genómico B.
Tabla 1 Porcentaje de identidad de nucleótidos calculados con el programa SDTv1.2 para el virus del mosaico dorado de Rhynchosia de Colombia aislado de cinco arvenses en el Valle del Cauca.
| Nombre del virus - acrónimo / número de accesión en GenBank | Hospedero | País | Porcentaje de identidad | ||
| Genoma A | Genoma B | ||||
| Rhynchosia golden mosaic Yucatan virus-RhGMYV / KP641347 - FJ792608 | Rhynchosia minima | México | 90,9 | 81,2 | |
| Rhynchosia rugose golden mosaic virus-RhRGMV / HM236371 | Rhynchosia minima | Cuba | ND | 80,2 | |
| Rhynchosia mosaic Sinaloa virus-RhMSV /DQ406672 - DQ356429 | Rhynchosia minima | México | 89,1 | 79 | |
| Tobacco yellow crinkle virus- TbYCV / KP641346 | Nicotiana tabacum | Cuba | ND | 78,8 | |
| Cabbage leaf curl Jamaica virus-CabLCJV / DQ178613 | Brasicaceae | Jamaica | ND | 75,8 | |
| Merremia mosaic Puerto Rico virus- MerMPRV / FJ944022 | Merremia quinquefolia | Puerto Rico | ND | 75,3 | |
| Capraria yellow spot Yucatan virus-CarYSYV/ KC426927 | Capraria biflora | México | 84,5 | ND | |
| Euphorbia mosaic Venezuela virus-EuMV / JN368145 | Euphorbia heterophylla | Venezuela | 83,2 | ND | |
| Tomato mild yellow leaf curl Aragua virus-ToMYLCV / AY927277 | Solanum lycopersicum | Venezuela | 82,8 | ND | |
| Euphorbia mosaic virus-EuMV / DQ318937 | Euphorbia heterophylla | México | 82 | ND | |
| Pepper golden mosaic virus-PepGMV/ LN848774 - LN848814 | Capsicum annuum | México | 81,9 | 74,5 | |
| Pepper huasteco yellow vein virus -PHYVV /LN848893 | Capsicum annuum | México | ND | 76,6 | |
| Euphorbia mosaic virus-EuMV / AF068642 | Euphorbia heterophylla | Puerto Rico | 81,7 | ND | |
| Potato yellow mosaic virus-PYMV / JQ045705 - JQ045706 | Solanum lycopersicum | Colombia | 74,1 | 67,3 | |
| Croton golden mosaic virus-CroGMV / KX156613 - KX156614 | Croton hirtus | Colombia | 73,9 | 69,3 | |
| Passionfruit leaf distortion virus-PDLV / KT899302 - KT899303 | Passiflora edulis | Colombia | 73,8 | 71,2 | |
ND. No determinado.
Análisis de los componentes genómicos A y B de RhGMCV
El alineamiento de la RI de los componentes genómicos begomovirales A y B reveló una región común de 190 nt con un valor de identidad de 87,7 %; por lo tanto, no se podrían considerar pares virales en la naturaleza pues el umbral para esto corresponde a identidad mayor a 90 % según se ha reportado (Brown et al., 2005). Para buscar la presencia de otro componente B se realizó la secuenciación del producto de PCR del fragmento de 0,5 kb amplificado de los aislados virales de las dos muestras de R. minima (AT35) y (AT36), encontrándose que ambos eran idénticos. Adicionalmente, cuando se clonó el genoma completo del begomovirus presente en R. minima (AT35), se obtuvieron cinco clonas que fueron secuenciadas las cuáles fueron entre sí idénticas. Con base en estos análisis no fue posible encontrar otro componente genómico begomoviral B cuya homología con el componente genómico A presente una homología superior al 90 %. Adicionalmente, al realizar el análisis a nivel de nucleótidos del componente genómico B contra otros componentes genómicos begomovirales presentes en las bases de datos, este presentó la mayor identidad (81,2 %) con RhGMYV (FJ792608] y 80,2 % con el virus del mosaico rugoso dorado de Rhynchosia (RhRGMV, HM236371), donde ambos virus infectan a R. minima en México y Cuba. Por otro lado, en campo se observó sintomatología viral en todas las arvenses de las cuales se aisló este virus: R. minima, R. humilis, Desmodium sp., Caesalpinia sp., y A. dubius, lo que podría indicar que el componente B identificado a pesar de no tener un porcentaje de identidad alto con su componente genómico A de alguna manera logra complementarlo, alcanzando este begomovirus su plena capacidad de diseminación a través de las arvenses en donde se encontró, cumpliendo así su ciclo infectivo. También está bien documentado que, si bien la presencia de ambos componentes genómicos A y B de un begomovirus particular permite a este género viral cumplir a satisfacción su ciclo infectivo, no es una condición sin excepción, ya hay evidencias de que ORFs del componente A en sí mismos actúan como factores de patogenicidad y determinantes de movimiento a través de las plantas aún en ausencia de su propio componente B.
También está muy bien documentado el fenómeno de trans-replicación entre begomovirus en donde un componente genómico begomoviral A puede complementarse, a nivel de la replicación, bajo ciertas condiciones, con otro componente begomoviral B a pesar de que este último no sea su 'compañero ' natural (Rojas et al., 2005; Ramesh et al., 2017). Entre las condiciones necesarias para que haya trans-replicación entre dos componentes genómicos begomovirales Ay B que no son 'compañeros' naturales se encuentran aquellas relacionadas con la presencia de ciertos elementos cis-regulatorios comunes en ambas RI presentes en ambos componentes. Fundamentados en esto, se llevó a cabo un análisis de los elementos cis -regulatorios presentes en RI de ambos componentes genómicos begomovirales de RhGMCV obtenidos en este estudio.
Los genomas A y B de RhGMCV presentan idénticos elementos cis-regulatorios en la región intergénica
Para verificar si es el componente B presenta en su RI elementos de secuencia comunes con aquellos presentes en la RI del componente genómico A, se realizó un análisis de secuencia en busca de elementos cis- regulatorios, encontrándose que ambos componentes presentan elementos típicos virales idénticos (Fig. 2). Por ejemplo, presencia de una secuencia conservada típica de la caja TATA, localizada río arriba del probable sitio de iniciación de la transcripción. Asimismo la presencia de una secuencia nonanucleotídica funcional (cuyo consenso es TAATATT*AC) la cuál es esencial para el inicio de la replicación y que forma que forma una estructura de tallo-asa (Hanley-Bowdoin et al., 1999)Adicionalmente se lograron identificar y localizar otros elementos de secuencia denominados iterones, los cuáles son claves no solo para la replicación geminiviral sino para que fenómenos como la trans-replicación entre componentes genómicos begomovirales puedan verificarse. Al respecto de esto en los genomas A y B de RhGMCV se encontraron cinco iterones típicos organizados de la siguiente manera: tres iterones en sentido directo cuya secuencia consenso fue 5' TGGAGTCC 3'; dos de estos iterones en sentido directo fueron ubicados dos ubicadas río arriba (hacia 5') de la caja TATA del promotor del gen de la proteína asociada a la replicación (Rep), mientras que el último de éstos iterones, en sentido directo, fue localizado río abajo (hacia 3') de la estructura de tallo y asa. Los restantes dos iterones cuya secuencia consenso fue 5' GGACTCCA 3' se encontraron en sentido invertido, aproximadamente 50 nt río arriba de la caja TATA del promotor del gen de la proteína Rep (Fig. 2, flechas azules). Estos análisis apoyarían nuestra hipótesis de que ambos genomas A y B de RhGMCV son pares virales, a pesar de presentar un porcentaje menor de 90 a nivel de identidad en su región común.

Figura 2 Alineamiento de secuencias de la región común de genoma del virus del mosaico dorado de Rhynchosia de Colombia (RhGMCV) con otros begomovirus que comparten la secuencia de iterones (señalada en azul), y los cambios en iterones con respecto a RhGMCV se resalta en rojo. La dirección de las flechas corresponde a la dirección de los iterones. La caja TATA se destaca en negro y la secuencia invariante de nona nucleótidos en vinotinto. El codón de inicio de la proteína de movimiento (MP) se indica en verde.
Adicionalmente, se realizó una comparación de interones en otras RI de begomovirus, encontrándose que RhGMCV pondría transreplicarse con ChLCV, RhGMYV, CarYSYV, EuMV y ToMYLCV, dado que presentan idénticos iterones en número y disposición. En la Fig. 2 se resalta la identidad total de iterones de los aislados con los virus de Mesoamérica, el Caribe y el suroriente de Estados Unidos e incluso con los virus aislados en Venezuela como el virus de la hoja rizado amarillo del tomate leve de Aragua (ToMYLCV) y el virus del mosaico de Euphorbia (EuMV). Se observan algunas variaciones en los iterones con el virus del mosaico dorado del chile (PepGMV), virus de la hoja rizada leve del Squash (SMLCuV), el virus de la hoja enrollada severa del tomate (ToSLCV) y algunos de los iterones de virus aislados de Euphorbia, a su vez presentan ligeros cambios.
En cuanto a la organización de iterones de RhGMCV, este se relaciona con el Clado del virus del encrespamiento de la hoja del squash (SqLCV) que tiene arreglo de iterones del Viejo Mundo más uno adicional a la región palindrómica (Argüello-Astorga et al., 1994)whose organization (number, orientation, and spacing que predomina en la región del Caribe y Mesoamérica. Por otro lado el virus de la hoja encrespada del calabacin (SqLCVE) tiende a conservar ligeramente cuatro de los cinco iterones de RhGMCV y dicha conservación ancestral de iterones puede estar vinculada a que lo que se afirma del origen de este, que SQLCVE emergió en América como resultado de un evento recombinatorio entre un begomovirus americano y geminivirus perteneciente a otro género (Argüello-Astorga et al., 1994)whose organization (number, orientation, and spacing. Este análisis nos permite postular que RhGMCV tienen altas probabilidades de pseudo-recombinarse con los virus de su Clado anotados en la Fig. 2 porque la semejanza en encontradas a nivel de secuencias en cis necesarias para la replicación hace de estos elementos de secuencia claves para que procesos como la trans-replicación entre componentes genómicos begomovirales pueda verificarse.
Relaciones filogenéticas de RhGMCV
El componente genómico A de RhGMCV se relaciona estrechamente con RhGMYV (KP641347) con quien comparte una identidad de 90,9 %. El componente genómico B de RhGMCV presenta una identidad de 81,2 % con RhGMYV (KP641347], RhGMYV (KP641347) afecta R. minima en México y se ubica en uno de los tres subclados de SLMV de virus principalmente distribuidos en México, como también de virus de Jamaica, Cuba y México (Tabla 1).
Las relaciones filogenéticas evidenciadas entre los begomovirus que infectan plantas cultivadas y aquellos presentes arvenses confirma el importante rol evolutivo que tienen las plantas acompañantes, como hospederos naturales de tales virus, así como el de servir de escenario en el cuál nuevos begomovirus pueden emerger gracias a mecanismos como la recombinación, la pseudo-recombinación, la trans-replicación y la trans-complementación.
Por tanto, podría considerarse la posibilidad de que los geminivirus que están en este momento afectando las plantas cultivadas a nivel mundial hayan evolucionado y emergido a partir de virus presentes en arvenses (Ambrozevicius et al., 2002; Barreto et al., 2013).
Las arvenses A. dubius, R. humilis, Desmodium sp. y Caesalpinia sp. presentan el virus del mosaico dorado de Rhynchosia de Colombia (RhGMCV)
La presencia de RhGMCV en las arvenses A. dubius (AT32), R. minima (AT35 y AT36), R. humilis (AT38), Desmodium sp. (AT40) y Caesalpinia sp. (AT41) se corroboró con un PCR específico que amplifica de un fragmento de 1,82 kb que porta los genes CP-TrAP-REn-Rep del componente genómico A de RhGMCV (Fig. 3). En el análisis de PCR, todas las muestras amplificaron un fragmento del tamaño esperado, confirmando la presencia de RhGMCV en las cinco arvenses evaluadas.
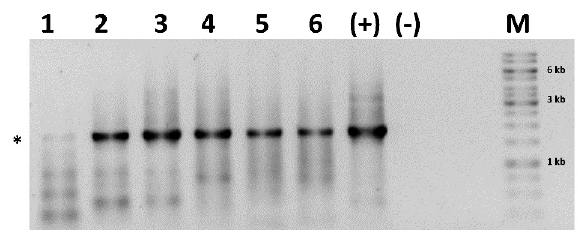
Figura 3 Confirmación de la presencia del begomovirus RhGMCV en cinco arvenses mediante la amplificación por PCR. Amplificación por PCR de un fragmento de 1,82 kb de RhGMCV-A. 1. Amaranthus dubius (AT32); 2. Rhynchosia minima (AT35); 3. Rhynchosia minima (AT36); 4. Rivina humilis (AT38); 5. Desmodium sp. (AT40); 6. Caesalpinia sp. (AT41); C+. Control positivo; C-. Control negativo. Gel de agarosa 1,0 %. M, Marcador de peso molecular gen ruler 1 kb DNA ladder Thermo Scientific. El asterisco indica el tamaño de 1,82 kb esperado.

Figura 4 Relaciones evolutivas de los genomas A y B del begomovirus RhGMCV aislado de Rhynchosia minima en Valle del Cauca. La historia evolutiva fue inferida usando el método de Neighbor-Joining (Saitou y Nei, 1987). El porcentaje de réplicas del árbol en el que los taxa fueron agrupados en un test de bootstrap (1000 réplicas). El árbol fue dibujado a escala, con el largo de las ramas en las unidades de las distancias evolutivas usadas para inferir el árbol filogenético. El virus del mosaico de yuca de África (Africa cassava mosaic virus -ACMV) se utilizó como grupo externo. Para nombre y nomenclatura de los virus utilizados ver Tabla 1.
CONCLUSIONES
Se reporta la obtención del genoma completo de un begomovirus bipartita aislado de las arvenses R. minima, R. humilis, Desmodium sp., A. dubius y Caesalpinia sp., el cual presenta dos componentes, A y B con un tamaño de 2 584 y 2 543 nt, respectivamente.
Teniendo en cuenta el actual criterio taxonómico del ICTV, éste Begomovirus bipartita presente en cinco arvenses en cultivos de tomate del Suroriente del Valle del Cauca es nuevo y se propone el nombre de Virus del mosaico dorado de Rhynchosia de Colombia (Rhynchosia golden mosaic Colombia virus, RhGMCV).
Nuestros resultados confirman el papel de las arvenses como hospederos alternativos de Begomovirus y como escenarios en donde nuevos virus de este género pueden emerger gracias a procesos de recombinación y pseudo-recombinación que a la postre conduzcan a la aparición de begomovirus mas patogénicos con altas y nuevas capacidades para adaptarse y ocupar nuevos hospederos, convirtiéndose así en limitantes para la producción agrícola a futuro.
Desde este punto de vista la epidemiología de los begomovirus que limitan la producción del tomate y otras dicotiledóneas de interés agrícola en Colombia estaría incompleto sin que se continúe abordando la Virosfera de este género geminiviral presente en las plantas acompañantes que rodean estos cultivos. Son los resultados del estudio de esta Virosfera los que se tomarán como base a futuro para establecer programas de manejo eficaces para contrarrestar la incidencia de las enfermedades begomovirales que limitan la producción agrícola en Colombia.


