











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
INTRODUCCIÓN
La investigación de fauna en islas continúa siendo un sistema informativo para comprender el efecto del aislamiento en la acumulación diferencial de biodiversidad (Valente et al., 2020). Las islas pueden funcionar como reservorios geográficamente discontinuos de poblaciones con características únicas o poco comunes en relación con sus pares continentales. En los sistemas insulares la discontinuidad geográfica respecto a sus áreas continentales es en sí misma, una hipótesis básica de la distribución espacial de la variación entre poblaciones (Templeton, 1989; Milá et al., 2010). Este tipo de hipótesis corresponde al ejercicio disciplinar de la filogeografía y aunque no se disputa lo indispensable de los análisis de secuenciación de amplia cobertura genómica, la información de secuencias parciales de ADN mitocondrial (ADNmt) es una fuente de datos con un marco analítico sólido (Zink y Barrowclough, 2008). Por esto, no es sorprendente la actualidad del uso de secuencias parciales ADNmt en un contexto filogeográfico de las mariposas en sistemas insulares o discontinuidades geográficas (Linares et al., 2009; Halbritter et al., 2019; Maresova et al., 2019).
Las mariposas constituyen un modelo interesante para el estudio de patrones de variación en el marco de discontinuidades geográficas (Brower, 1994; Todisco et al., 2010). A pesar de su aparente fragilidad fenotípica, las mariposas, han logrado colonizar y segregarse genéticamente en remotas islas (Miyakawa et al., 2018), pero este no siempre es el caso en condiciones de presumible aislamiento (Halbritter et al., 2019). Esto sugiere que los procesos relacionados con aislamiento son complejos y requieren aún ser documentados. Las islas a menudo constituyen áreas marginales del rango de distribución de algunas poblaciones de mariposas, que, a su vez, podrían ser particularmente susceptibles a factores de variabilidad climática (Descimon, 1995). Estudios filogeográficos documentan importantes detalles sobre el patrón de ancestría y descendencia tanto de grupos en amplios rangos, como de aspectos de patrones demográficos a pequeña escala espacial (Todisco et al., 2010; Maresova et al., 2019).
En América, la distribución geográfica de mariposas continúa siendo documentada como un sistema excelente para comprender patrones de variación (Chazot et al., 2018). Aunque la literatura sobre la evolución del género Heliconius es prolífica y muestra el contexto filogenético de atributos de su historia natural (Montejo-Kovacevich et al., 2019), su sistemática (Kozak et al., 2015) incluyendo patrones introgresión (Zhang et al., 2016) y convergencia entre genomas (Morris et al., 2020), como también de patrones de divergencia, flujo de genes (Nadeau et al., 2013) y evidencia de selección natural (Martin et al., 2016). Para la mayoría de las especies del género no existe un esfuerzo equivalente, esto a pesar de que existe una asombrosa colección de la variación de su apariencia externa. Algunos estudios muestran un comprensivo esfuerzo por caracterizar la filogeografía de algunas especies en Heliconius (Arias et al., 2014) y la diferenciación entre poblaciones geográficamente delimitadas parece ser un patrón común en Heliconius (de Moura et al., 2011). Otros estudios ofrecen evidencia de divergencia dado el contexto vicariante que ofrecen los sistemas de montañas como los Andes (Brower, 1994).
Algunos estudios que consideran la distribución espacial de la variación genética en este género han demostrado inferir correctamente la relación de ancestría y descendencia entre algunas especies con marcadores del ADNmt (Brower, 1994; Pardo-Diaz et al., 2012; Arias et al., 2014). Así mismo, este tipo de marcador genético es documentado como informativo para realizar estimaciones de reloj molecular en estas mariposas (Brower, 1994; Arias et al., 2014). Lo anterior sugiere que, los fragmentos de ADNmt continúan proporcionando información de la distribución espacial y de la variación genética neutral como una aproximación preliminar en el cálculo de la distancia entre poblaciones.
Heliconius sara (Fabricius, 1793), es un lepidóptero típico en hábitats continentales e insulares de Centro y Sur América. Esta mariposa de alas elongadas, común en áreas abiertas del bosque, es una especie para la cual se han documentado hábitos gregarios y especialistas de plantas hospederas de la familia Passifloraceae (Ehrlich y Raven, 1964; DeVries, 1987). En esta especie no existe un dimorfismo sexual marcado en su morfología externa; su apariencia tiene un carácter aposemático con bandas de coloración amarilla en el ala sobre un fondo melánico de visos en azul metalizado. Un patrón de coloración de apariencia mimética con otras especies resultado de convergencia no-estructural (Thurman y Seymoure, 2016; Parnell et al., 2018). Aunque H. sara presenta polimorfismo en el patrón de coloración entre una decena de subespecies, en general este polimorfismo representa una morfología conservada con relación a especies con las que comparte un ancestro común cercano en el subclado Sapho-Sara (Kozak et al., 2015).
Se ha propuesto una baja capacidad de dispersión al menos en una forma de Heliconius dentro de su rango de residencia habitacional; de acuerdo con Mallet (1986), aún se requieren avances en el conocimiento del potencial de dispersión de las especies en este género. La capacidad de dispersión de H. sara aún se desconoce y aunque su rango de distribución alcanza la Isla Gorgona, a 35 km de la costa del suroccidente de Colombia, en la región del Pacifico oriental tropical (Calero-Mejía et al., 2014). Otras especies comunes del género en la región del Chocó Biogeográfico en el Pacifico colombiano, no están presentes en los registros de los lepidópteros diurnos en esta isla. De esta observación, es razonable suponer capacidades diferenciales interespecíficas en el potencial de dispersión, colonización o extinción en ambientes insulares y potencialmente en el patrón espacial de la distribución de la variación genética.
En la Isla Gorgona se ha documentado la acumulación de endemismo en diferentes grupos biológicos (Fowler 1994; Giraldo et al., 2014), un patrón espacial de la diversidad que aún debe ser examinado sistemáticamente para mariposas y otros insectos terrestres. Por lo tanto, es razonable suponer que, presumiblemente el rango de H. sara en la isla Gorgona separada de la costa del Pacifico colombiano por la actual discontinuidad marítima, podría ser congruente con el patrón de endemismo observado en esta isla.
El objetivo del presente trabajo fue caracterizar la variación genética de la población que reside en la Isla Gorgona en un contexto filogenético explícito, que busca examinar la presumible monofilia mitocondrial de H. sara, mediante el análisis de secuencias parciales del ADNmt para el gen Citocromo oxidasa I (COI). Con base en este supuesto, se analizó el potencial aislamiento de la población insular desde una perspectiva filogeográfica y se examinaron modelos de migración histórica. La información obtenida podría contribuir al entendimiento del fenómeno de dispersión, y a la caracterización de poblaciones de mariposas diurnas como objetos de conservación en sistemas insulares de Colombia.
MATERIALES Y MÉTODOS
Área y objeto de estudio
El área de interés para este estudio fue el Parque Nacional Natural Isla Gorgona (2°9' N y 78°2' W). La isla presenta una cobertura de selva húmeda en una geografía que alcanza una elevación máxima de 338 m. El clima es cálido con temperatura promedio de 26 °C (Rangel-Ch., 1995) y se ha documentado una pluviosidad media anual de 6981 mm, que supera en precipitación a las áreas continentales cercanas como la localidad de Guapi a ca. 60 km (Blanco et al., 2009). Para caracterizar la población de H. sara que habita en la Isla Gorgona, se colectaron especímenes con red entomológica durante caminatas por transectos de longitud no definida. Los especímenes colectados fueron inmovilizados mediante presión torácica, luego puestos en una cámara con acetona. Para la preservación de las alas se guardaron en sobres de papel encerado, por último, utilizando alfileres, se montaron con las alas extendidas en cajas depositadas en el Museo de Entomología de la Universidad del Valle (MUSENUV).
Extracción, PCR y Secuenciación de ADN
Para la extracción de ADN total se empleó un par de patas de cada espécimen de H. Sara. Las patas fueron sometidas a una solución de lisis a 56 °C durante 12 h en agitación constante. La extracción se realizó con el kit comercial ®Thermo Fisher Scientific siguiendo el protocolo del fabricante. La muestra de las patas de cada espécimen y extracciones de ADN total, se almacenaron a -20 °C para su conservación. Para la amplificación por la PCR del ADNmt del gen COI se usaron los cebadores CAACATTTATTTTG ATTTTTTGG (Jerry-COIF) y GCTACTACATAATAKGTATCATG (Ben-COIR) (Simon et al., 1994). En la mezcla de la PCR se utilizó 3,5 μl de H2O, 12,5 μl de Taq DNA polimerasa (1X One Taq Hot Start Quic Maxter Mix), 2,5 μl BSA, 0,5 μl DMSO, 0,5 μl de cada cebador, 5 ng de DNA para un volumen total de 25 μl. La amplificación se realizó en un Termociclador Benchmark TC9639 con una denaturación inicial de 94 °C (4 min), seguido de una más corta a 94 °C (40 s), 45 °C (1 min) de alineación, una elongación a 72 °C (1,5 min) por seis ciclos desde el primer paso. Luego se realizó una segunda fase de denaturación a 94 °C (1 min), alineación de 55 °C (1 min), una elongación a 72 °C (1,5 min) por 30 ciclos desde el inicio de la segunda fase y una extensión final de 72 °C (10 min) seguida de 10 °C (5 min). Los productos de la PCR fueron separados por electroforesis en un gel de agarosa de 2 %. Las muestras fueron secuenciadas por el servicio de MACROGEN Inc. Las secuencias obtenidas fueron editadas y alineadas visualmente con el programa Sequencher 4.1.4 (Gene Codes Corporation, Ann Arbor, Michigan, USA). Para esto se corroboró la identidad de las secuencias usando la plataforma BLAST (Johnson et al., 2008) y todas las secuencias de novo se encuentran disponibles en el GenBank (Benson et al., 2011) (Tabla 1).
Tabla 1 Accesión de secuencias del GenBank para las unidades taxonomicas operativas seleccionadas del subclado Sapho-Sara, además del país y localidad de origen.
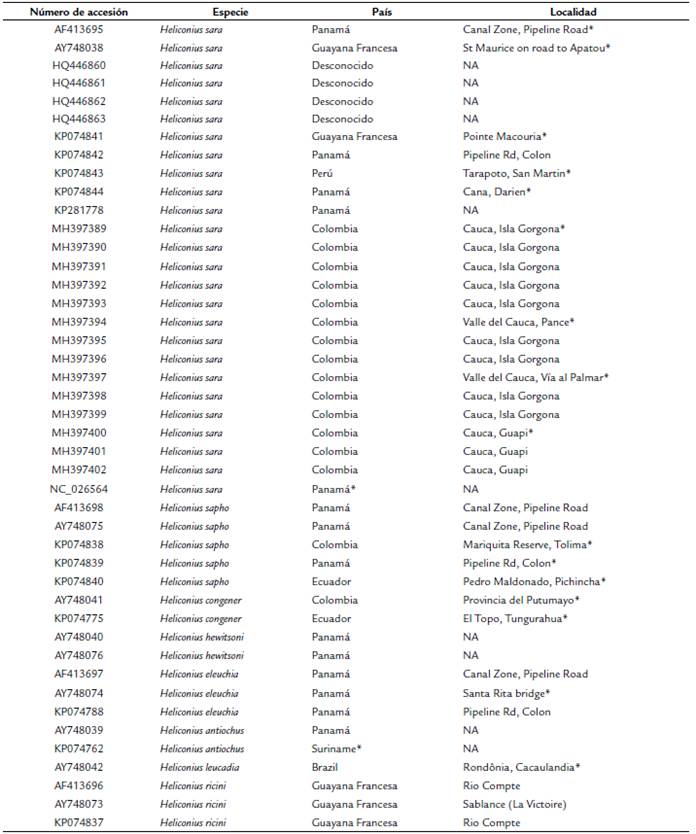
*Localidades usadas para realizar la prueba de Mantel (ver métodos). NA indica provincia indeterminada.
Para tener una mejor comprensión acerca de la diversidad genética de H. sara y en particular de la población en la Isla Gorgona, primero se examinó un contexto filogenético más amplio y se incluyeron algunas Unidades Taxonómicas Operativas (UTOs) pertenecientes al subclado Sapho-Sara (Kozak et al., 2015): H. antiochus (Linnaeus, 1767), H. leucadia (Bates, 1862), H. hewitsoni (Hewitson, 1875), H. sapho (Drury, 1782), H. congener (Weymer, 1890) y H. eleuchia (Hewitson, 1854) (ver Tabla 1). El mapa de distribución de los especímenes georreferenciados en este estudio se realizó en QGis (QGIS.org, 2020).
Diversidad nucleotídica
Con el alineamiento de las secuencias parciales del gen COI se calcularon los sitios variables (S), la diversidad nucleotídica (n), así como el estadístico D de Tajima para aquellas unidades cuando el tamaño de muestra fue adecuado, y para esto se usó el programa Arlequín 3.5.2.2 (Excoffier y Lischer 2010). Con este alineamiento también se realizó una determinación de haplogrupos, definidos aquí con un criterio de mínima identidad nucleotídica, que resultará en la discriminación máxima entre H. sara, H. eleuchia, H. congener, H. sapho, H. hewitsoni, H. antiochus, H. leucadia usando para esto el algoritmo USEARCH (Edgar, 2010).
Estructura Genética
Se realizó un análisis de varianza molecular (AMOVA) para comparar la población de H. sara de la Isla Gorgona y sus conespecíficos continentales usando el programa Arlequín 3.5.2.2 (Excoffier y Lischer 2010). Con el AMOVA se calculó el índice de fijación Φst, con una prueba exacta de diferenciación entre UTOs realizando 16 000 permutaciones. Como complemento al AMOVA y para examinar la relación espacial de las distancias genéticas entre localidades, se calculó el patrón espacial de variación interespecífica con un modelo de aislamiento por distancia (APD). Para el análisis de APD la matriz de distancia genética patrística entre UTOs fue calculada con MEGA X (Kumar et al., 2018) y la distancia geográfica fue obtenida con una calculadora en línea (ver "Geographic Distance Matrix Generator" por (Ersts, 2014). Con estas matrices se realizó una prueba de Mantel con el paquete Vegan en RStudio Team (2016) (RStudio, Inc., Boston, MA, USA) entre 18 localidades de Centroamérica y Suramérica en donde se han reportado estás subespecies en la literatura citada (Beltrán et al., 2002; 2007; Kozak et al., 2015) y el GenBank (Benson et al., 2011). Para visualizar el patrón de asociación más parsimoniosa entre haplogrupos, se construyó una red entre estos utilizando valores de la mediana entre haplogrupos para su unión en red con el programa PopArt v.1.7 (Leigh y Bryant, 2015).
Genealogía por Máxima Verosimilitud y por Inferencia Bayesiana
La reconstrucción de la genealogía se realizó por el método de Máxima Verosimilitud (MV) en la plataforma de PhyML 3.0 (Guindon et al., 2010) y se ejecutó sin restricción alguna sobre la topología. El árbol inicial presenta puntas aleatoriamente distribuidas y se incluye a H. ricinicomo grupo externo para UTOs aquí seleccionadas dentro del subclado Sapho-Sara. Se implemento para el análisis a un modelo de sustitución HKY, ajustado con igual frecuencia para todas las bases. Este modelo con un valor de heterogeneidad en el patrón de sustituciones dividido en diez categorías Gama, con una distribución normal para el parámetro de transiciones y transversiones que tienen un valor inicial de 0 y una media de 3,14625 (sd = 1,7). Se determinaron a priori dos particiones para las posiciones en el codón (1+2 y 3). Al final se realizó un consenso con 5000 repeticiones para calcular porcentualmente el soporte de los nodos.
La genealogía fue calibrada con los aspectos mejor soportados de la topología del subclado Sapho-Sara de acuerdo con Kozak et al. (2015). En esta filogenia se documenta un alto soporte para un clado conformado por H. sapho, H. eleuchia y H. congener. El segundo grupo restringido incluye todas las UTOs en este estudio como un grupo monofilético, pero excluyendo a H. ricini (ver Tabla 1). Este patrón de la topología fue ajustado en BEAUTi v1.8.4 (Drummond et al., 2012), la reconstrucción de la genealogía se realizó con el método de IB en BEAST v1.8.4 (Drummond et al., 2012) como esta desplegado en la plataforma CIPRES (Miller et al., 2010). Para este análisis se implemento el mismo modelo del análisis por MV, misma partición de datos, igual patrón de sustituciones con diez categorías y se estableció una longitud de cadena de 10 000 000 pasos, recolectando datos cada 1000 pasos. El reloj molecular se indicó de carácter relajado Log normal con un modelo coalescente de tamaño poblacional constante. El ancestro común más cercano al grupo interno fue calibrado con una media de 2,1 ±1,0 millones de años de acuerdo con Kozak et al. (2015). Con el anotador de árboles TreeAnnotator v1.8.4 (Drummond et al., 2012), se procesó el archivo de salida producido en BEAST v1.8.4 y se excluyó un exceso del 25 % de las inferencias iniciales (Burn-in). Con los datos restantes se generó un árbol de mayor credibilidad y su visualización se realizó en el programa MrEnt v2.5 (Zuccon y Zuccon, 2014). La congruencia entre los resultados por IB y MV se examinaron usando como referencia el soporte de los nodos mayor o igual a 0,95 para la IB y al 70 % para el análisis de MV.
Análisis demográfico para H. sara
Con el fin de examinar el patrón histórico de migración para H. sara entre el área continental y la Isla Gorgona, se realizó un análisis de la coalescencia del marcador COI. Con este alineamiento de secuencias se estima de manera conjunta el tamaño efectivo poblacional (θ) y la tasa de migración total (M) (ambos escalados a la tasa de mutación) mediante un método de IB en Migrate-n 4.2.14 (Beerli et al., 2019). Para este análisis se utilizó el modelo de sustitución nucleotídica HKY con el objetivo de realizar un análisis consistente con el modelo implementado para el método de inferencia de la genealogía por IB. En Migrate-n se programó una cadena de 250 000 pasos, Burn-in de 200 000 pasos iniciales, con 20 repeticiones independientes del análisis para calcular un promedio de la estimación de los parámetros. Adicionalmente, se implementó un modo adaptativo de muestreo durante la IB en Migrate-n, donde a cada paso se realizan intercambios de aceptación y rechazo en las estimaciones, lo que facilita el rastreo de los parámetros sobre el paisaje estadístico. La estacionalidad del proceso de la IB fue determinada por los valores ESS >104para cada parámetro. La aproximación de Bézier de integración termodinámica (implementada en Migrate-n) fue utilizada para estimar el valor marginal de verosimilitud y con este valor calcular la probabilidad condicionada a los datos de los diferentes modelos de migración (Apéndice 3). Con el modelo de mayor probabilidad se reinterpretan los valores de M, como el cálculo en RStudio Team (2016) de un producto con θ que se aproxima al número de inmigrantes efectivos por generación (Nm) (Beerli et al., 2019).
RESULTADOS
Variación Genética
Se obtuvieron 14 secuencias del gen COI de especímenes de H. sara para Colombia, de los cuales nueve provienen de la isla Gorgona (Fig. 1). Con la consulta realizada en el GenBank se obtuvieron 35 secuencias adicionales de áreas continentales, tanto para H. sara (17 secuencias) como para las demás especies del subclado Sapho-Sara (18 secuencias). Estas últimas incluidas para efectos de examinar la hipótesis de monofilia de H. sara. Un total de 44 accesiones de secuencias de COI fueron alineadas de modo que presentan información de un fragmento de 508 pares de bases para el gen COI (Tabla 1). El alineamiento presenta menos del 1,2 % de datos faltantes, un 77,6 % de loci invariables, mientras que el 19,1 % corresponde a loci informativos. La definición de haplogrupos con un criterio de similaridad nucleotídica > 98 % entre secuencias parciales del gen COI en USEARCH es congruente con el resultado de la red de haplogrupos. Esto implica que en la red de haplogrupos un mínimo de 2 % de diferenciación genética permite discriminar entre todas y cada una de las UTOs (Fig. 2a).

Figura 1 Morfología externa de Heliconius sara de la Isla Gorgona, Colombia. En vista ventral (Izq) los adultos son negros opacos con manchas rojas en la zona basal y postbasal del ala posterior. El ala anterior en vista ventral tiene una franja clara en la zona de traslape con el ala posterior, presenta una línea amarilla en el margen costal del ala anterior desde la base hasta la zona media y dos manchas de color amarillo claro en la zona medial y apical de las alas. En la vista dorsal (Der.) se aprecia que el color negro es más intenso y se mezcla con las tonalidades de un azul iridiscente que se extiende desde la base hacia las márgenes distales. En vista dorsal son ausentes las manchas rojas observadas en vista ventral, pero las dos bandas del AA se conservan, contrastando con el negro en una señal aposemática.
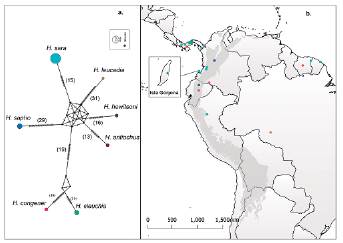
Figura 2 a) Red de haplogrupos de especies del subclado Sapho-Sara incluidas en este estudio. El tamaño de los círculos es proporcional al número de secuencias disponibles de cada haplogrupo en el GenBank. Las líneas entre los haplogrupos y los números entre paréntesis representan el número de pasos mutacionales. La estructura geométrica donde convergen todas las especies indica una politomía dado que este análisis no está polarizado con el grupo externo en el subclado Sapho-Sara (ver Métodos). b) Distribución de las localidades de donde provienen las secuencias reportadas en GenBank de las especies objeto de análisis con un recuadro de ampliación para ilustrar la localidad Isla Gorgona (ver Métodos y Tabla 1).
Análisis de Varianza Molecular
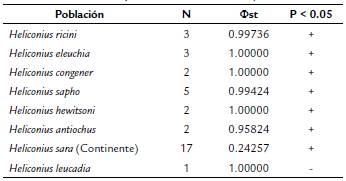
La distribución del patrón de variación nucleotídica del gen COI es diferencial entre y dentro de las UTOs del subclado Sapho-Sara [AMOVA Φst 835 = 0,94483, prueba exacta de distribución individual P = 0,00001]. Esto indica que la diferenciación promedio entre UTOs es mucho más alta (Φst) que el mínimo valor (2 %) de diferenciación calculado en el análisis con USEARCH. Considerando el tamaño de la muestra para cada UTO, la prueba de Tajimas D solo fue posible calcularla para H. sara y H. sapho. Los valores negativos del índice D sugieren que el conjunto de datos para estas dos formas de Heliconius podría presentar un exceso de variantes de menor frecuencia; sin embargo, la hipótesis de neutralidad de la variación en COI no puede ser rechazada para H. sapho (D= -0,81650, p= 0,30500) y H. sara (D=-1,51978, p= 0,0520).
Existe evidencia, de un patrón de diferenciación entre la población de H. sara recolectada de la Isla Gorgona con su segmento conespecífico de distribución en el área continental (ver Tabla 3). Así como también, se encuentra un patrón de diferenciación genética para H. sara de la isla con las demás UTOs excepto con H. lecaudia con quien su valor Φst no puede ser diferenciado (ver Tabla 3). Sobre lo anterior se enfatiza que la estructura genética observada entre UTOs no puede ser explicada a priori por el patrón de distribución espacial de las muestras. Esto quiere decir que la variación genética entre especies del subclado Sapho-Sara presenta una muy pobre correlación lineal con la estructura de la matriz de distancias geográficas (r = -0,002425, p = 0,45455, perm=10000).
Tabla 2 Número de nucleótidos y tamaño de la muestra para calcular índices de diversidad molecular para la especie H. sara de la Isla Gorgona, del área continental y otras UTOs del subclado Sapho-Sara.
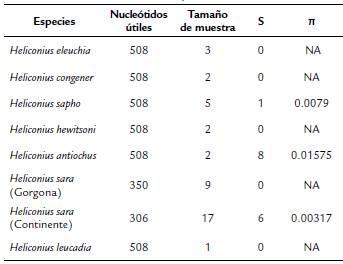
NA: no procede el cálculo dado que (i) el tamaño de la muestra es uno o (ii) S=0.
Inferencia filogenética
Se utilizó el alineamiento del conjunto de 44 accesiones de secuencias del gen COI, incluyendo el grupo externo del subclado, para la selección de un modelo de sustitución nucleotídica HKY (BIC= 3770, K=91) para mitigar el efecto de la potencial saturación de sustituciones en ese fragmento de ADNmt. Este modelo de sustitución y el proceso de inferencia filogenética por MV (LogMV = -1616,68932) es parcialmente congruente con la IB en la que se predeterminaron aspectos de la topología del clado Sapho-Sara (Ver Apéndice 1). Ambos análisis coinciden en el soporte del 56 % de los subclados en la genealogía resultante pero solo el análisis de remuestreo por MV ofrece un 93 % de soporte para la monofilia de las secuencias de H. sara (Apéndice 1). De acuerdo con la calibración de la genealogía por la IB, la diversificación haplotípica en H. sara es muy reciente, comenzó grosso modo en el Pleistoceno superior hacia principios del Holoceno entre 0,0796-0,0143 millones de años (Apéndice 1).
Análisis demográfico
El análisis del patrón histórico de migración de H. sara entre el área continental y la Isla Gorgona sugiere un ajuste razonable con un modelo que obtuvo el mayor valor de probabilidad 0,95 (Apéndices 2 y 3). El resultado soporta la conjetura de un patrón de estructura genética y rechaza la hipótesis nula de una población panmíctica. El modelo de tipo XX0X (como se parametriza en Migare-n) (Beerli et al., 2019), describe flujo genético asimétrico de H. sara y sugiere la posibilidad de una historia de migración unidireccional desde la isla hacia el continente (Apéndice 3). Para el modelo, la estimación de parámetros demográficos θ continente' θ Gorgona’ y M Gorgona-Continente ' ' 0,00002 y 217,5 respectivamente y curvas unimodales de distribución de frecuencia para la estimación de cada parámetro (Apéndice 2). Sin embargo, el intervalo del 95 % de confianza para los parámetros θ Gorgona (0,0-0,14228), θ continente (0,0-0,14228) y M Gorgona→Continente . (0,0 a 18150,0) presentan una parte de su curva de distribución de frecuencia que podría no diferenciarse claramente de cero, y por esto, la interpretación de los valores modales debe realizarse con precaución. Con esto en consideración, en el patrón histórico de migración de H. sara la conversión del número total de migrantes M a Nm resulta en ca. 18 inmigrantes por generación. Si la inferencia de los valores modales de los parámetros estimados es adecuada, entonces Nm, es el número de individuos que probablemente ha fluido históricamente por generación hacia áreas continentales desde la isla. Sin embargo, para efectos de la interpretación del modelo es importante considerar que la cantidad de variación en el alineamiento de secuencias parciales de COI podría ser insuficiente para la estimación de una distribución de M con todos sus valores diferentes de cero.
DISCUSIÓN
El contexto filogenético de H. sara
En este estudio se examina el patrón de variación genética de H. sara entre una población con distribución en la isla Gorgona y aquellas muestras de la misma especie con distribución en el área continental en un contexto de análisis inter-específico (Fig. 2b). Para esto se ha caracterizado un fragmento de ADNmt del gen COI que ha sido documentado como un marcador informativo en el estudio de ancestría y descendencia de mariposas, y en particular del género Heliconius (Linares et al., 2009; Arias et al., 2014). Este análisis implementa la inferencia por MV de una genealogía con un fragmento de ADNmt qué es consistente con la filogenia del clado Sapho-Sara de Kozak et al. (2015). La inferencia muestra evidencia de (i) soporte por MV para un grupo monofilético de secuencias de ADNmt de H. sara y (ii) la datación por IB acotada sobre la genealogía del clado de H. sara a partir de secuencias que provienen de un amplio marco geográfico.
La red de haplogrupos de las UTOs constituye una síntesis gráfica de la genealogía desde una perspectiva ultramétrica por el criterio de parsimonia y representa gráficamente que una distancia genética (no corregida) de un 2 % en promedio permite diferenciar mínimamente entre todos y cada uno de los diferentes UTOs (Fig. 2a). La diferenciación mínima representada en la red es consistente con la diferenciación porcentual entre especies del género Heliconius que se ha documentado con fragmentos aún más largos de ADNmt (Mallet et al., 2007). El análisis de ultrametrización de la genealogía mediante la IB calibra la datación como un fenómeno de diversificación reciente para la distribución diferencial de la variación nucleotídica de ADNmt de H. sara entre otras especies del género Heliconius. En adición, la evidencia de la monofilia de secuencias parciales de ADNmt de H. sara soporta el tratamiento de este subconjunto de datos en un marco de análisis demográfico.
Diversidad de H. sara
La diversidad nucleotídica observada en H. sara es menor a lo reportado para otras especies del género como H. melpomene (Linnaeus, 1758), H. cydno (Doubleday, 1847) y H. timareta Hewitson, 1867 (Arias et al., 2014) o que lo calculado aquí para H. Sapho (Dury, 1782). En Costa Rica se han documentado valores moderados de variación genética local de H. sara, en relación con otras especies del género en condiciones sintópicas (Kronforst y Gilbert, 2008). Para H. sara presentamos evidencia de un patrón de diferenciación genética consistente con el aislamiento de la población en la isla, presumiblemente asociado con la discontinuidad geográfica con el continente. La distancia genética entre poblaciones de H. sara en la isla y el continente es menor que la distancia entre H. sara con cualquiera de las otras especies incluidas en este estudio con las que comparte un ancestro común cercano (Kozak et al., 2015). Es importante enfatizar que la distancia genética entre H. sara entre la isla y el continente es más parecida al nivel de diferenciación intraespecífica observado en el género Heliconius (Arias et al., 2014).
Análisis demográfico
El análisis de la coalescencia soporta un modelo de flujo restringido hacia la isla como el patrón histórico de dispersión efectiva desde la isla hacia el continente. Una consideración importante en la interpretación de este análisis es que no es posible obtener información de la incertidumbre asociada a la estimación del proceso coalescente con ADNmt como el único locus analizado (Zink y Barrowclough, 2008). Sin embargo, el modelo obtenido con Migrate-n sugiere que las estimaciones de 6 son consistentes con el supuesto de un tamaño efectivo de la población menor en la isla comparado con el área continental (Apéndice 2). De modo que si la inferencia en Migrate-n del parámetro M es una aproximación cercanamente correcta, este análisis puede resultar más informativo del fenómeno de migración efectiva, que pueda ser interpretado a partir del cálculo del índice de fijación Φst (Bohonak y Roderick 2001). Así, el modelo aquí seleccionado sugiere un patrón histórico de abundante inmigración por generación (Nm) de la isla hacia el rango de la especie en el área continental del pacifico colombiano. Cabe aquí recordar un evento anecdótico importante entre los lepidópteros de la isla, en el que H. sara es una de las pocas especies que ha sido documentada volando en mar abierto y en dirección al continente (Calero-Mejía et al., 2014). Lo cual ofrece evidencia circunstancial del potencial de dispersión de esta mariposa sobre esta discontinuidad geográfica de 35 km en la actualidad.
Dado que se presume que, el fenómeno de hibridación con otras especies es particularmente raro en H. sara (Brown et al., 1992). Los resultados de diferenciación porcentual promedio observada entre la población del continente y la Isla Gorgona, así como el soporte de monofilia para el conjunto de secuencias parciales de ADNmt, sugiere mínimamente, una aminorada posibilidad de anticipar evidencia de herencia uniparental matrilineal como parte de un fenómeno de hibridación interespecífica con H. sara. Esto sería consistente con la predicción de cruces interespecíficos en el género Heliconius en condiciones silvestres (Mallet et al., 2007). Sobre los resultados aquí presentados se especula que, la historia coalescente de H. sara posiblemente no ha sido sustancialmente distorsionada por evolución reticulada (al menos no ligada a herencia de ADNmt). Sin embargo, la evolución reticulada es un fenómeno aparente entre especies más o menos distantes en su relación filogenética en el género (Zhang et al., 2016), como ha sido documentado en H. heurippa (Hewitson, 1854) (Salazar et al, 2008).
Por primera vez se examina el posible contexto de aislamiento de una población de mariposa en la Isla Gorgona, Colombia. Se utilizó en este estudio un segmento de ADNmt con el que se obtiene información sobre la distribución espacial de la variación genética neutral entre la isla y el área continental. Además de marcadores como el ADNmt que ha sido documentado como informativo para lepidópteros; otros marcadores de la determinación del sexo también se han documentado cómo prometedores para estudiar la historia demográfica en el género Heliconius (Van Belleghem et al., 2018).
Los resultados aquí presentados sugieren un patrón de diversificación reciente con dispersión restringida hacia la Isla Gorgona. Este parece un modelo histórico consistente con la distribución diferencial del patrón de variación del ADNmt de la población de H. sara entre la Isla Gorgona y el área continental. Otras fuentes de datos, como la variación fenotípica o el análisis de patrones de dispersión local que puedan ser implementados en estudios posteriores, podrían contribuir a establecer el grado de congruencia con el patrón aquí observado de acumulación de variación genética neutral en H. sara de la Isla Gorgona.
CONCLUSIONES
Este estudio es una contribución a la caracterización de poblaciones de lepidópteros diurnos en la región del Pacifico colombiano. La forma de H. sara en la isla presenta un patrón de diferenciación genética neutral característica de la diferenciación intraespecífica observada en el género Heliconius. Proponemos un modelo histórico relativamente simple de flujo de genes asimétrico y restringido del área continental hacia la isla que podría remontarse -como una estimación cautelosa- a los últimos 80 k años. Estudios en el futuro de secuenciación de amplia cobertura genómica en esta especie podrían examinar el modelo aquí presentado. Nuestro modelo soporta la noción de divergencia local en aislamiento para esta población insular, promovido por restricción en el flujo de genes. La restricción de flujo del continente hacia la isla se daría presumiblemente por la actual barrera marítima con el continente; evidencia anecdótica de H. sara sobrevolando la discontinuidad marina desde la isla da cuenta de su potencial de dispersión. Un esfuerzo de investigación complementario que incluya la observación de patrones de dispersión in situ podría contribuir a analizar el modelo histórico aquí presentado. Este modelo también puede ser utilizado en el estudio de otros aspectos de la biología en esta especie como la evolución reciente de características fenotípicas en ese contexto espacial (Fig. 1). La congruencia de nuestra hipótesis de trabajo para otras especies de mariposas diurnas e insectos en la isla con distribución en el área continental continua de Chocó Biogeográfico es una pregunta abierta que aún queda por resolver.

