











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
INTRODUCCIÓN
El cultivo de la papa criolla (Solanum phureja Juz. et Buk.) es uno de los renglones agrícolas más promisorios para la región Andina de Colombia y otros países suramericanos, gracias a su corto ciclo de producción (4-5 meses), ausencia de dormancia en sus tubérculos, excepcionales características organolépticas y altos niveles de carotenoides (i.e. xantofilas, neoxantinas, violaxantinas y luteínas), vitaminas B y C, y microelementos (Fe, Zn, Cu y Ca) (Rodríguez et al., 2009). La región del suroccidente de Colombia es un centro de biodiversidad de esta especie de papa y desde allí su cultivo se ha dispersado a diferentes departamentos del centro del país como Cundinamarca, Boyacá y Antioquia (Rodríguez et al., 2009; Navas y Díaz, 2012). Para 2017 la producción total de papa criolla en Colombia fue de aproximadamente 130 000 t en un área de siembra de 8 460 ha, con rendimientos promedio de 14,2 t/ha; aunque dicho valor presenta grandes variaciones entre departamentos. De esta forma, en Norte de Santander y Cundinamarca se registraron rendimientos de 21,07 y 17,93 t/ha, respectivamente, mientras que en Antioquia este valor fue de tan sólo de 12,11 t/ha (Agronet, 2019).
En Antioquia, la papa criolla generalmente se siembra en lotes de menos de dos ha y se cultiva por pequeños agricultores que recurren a su siembra como fuente de alimento para sus familias. Estos, además, utilizan los excedentes como tubérculos-semilla para el siguiente ciclo y para su comercialización en mercados locales o regionales (Navas y Díaz, 2012). La siembra de la papa criolla ocurre de manera intensiva en dos a tres ciclos, seguidos por rotaciones frecuentes con pastos, leguminosas e incluso con otras plantas solanáceas como uchuva (Physalis peruviana L.) y tomate de árbol (Solanum betaceum (Cav.) Sendt.). Algunas de las principales limitantes para la producción de papa criolla son el tizón tardío (Phytophthora infestans (Mont.) de Bary), la sarna común (Streptomyces scabies Lambert and Loria), la sarna polvosa (Spongospora subterranea (Wallr.) Lagerh) y diversas enfermedades de origen viral (Navas y Díaz, 2012). Los agentes causales de las enfermedades virales hasta ahora detectados en este cultivo en Antioquia incluyen Potato leafroll virus (PLRV), Potato mop-top virus (PMTV), Potato virus S (PVS), Potato virus V (PVV), Potato virus X (PVX), Potato virus Y (PVY) y Potato yellow vein virus (PYVV) (Sánchez et al., 1991; Gil et al., 2011, 2012, 2013; Guzmán et al., 2012; Villamil-Garzón et al., 2014; Alvarez et al., 2016; Gutiérrez et al., 2016; Mesa et al., 2016; Vallejo et al., 2016; Gallo et al., 2019).
Estudios recientes han demostrado que uno de los aspectos que más afectan la sanidad de los cultivos de papa en los países en desarrollo, corresponde a la degeneración de los tubérculos-semilla, dada la ausencia de programas sostenidos de certificación de semilla (Thomas-Sharma et al., 2016; Gallo et al., 2019). Desafortunadamente, en Colombia es muy limitada la oferta de semilla certificada de papa criolla y esto se ve reflejado en los altos niveles de infección viral que se presentan tanto en el material de siembra como en los cultivos. En este sentido, Sierra et al. (2020) evaluaron mediante RT-qPCR los porcentajes de prevalencia de seis virus de ARN en tubérculos-semilla de papa criolla comercializados en Antioquia. Los autores encontraron niveles muy altos de infección para todos ellos (> 40 %), lo que confirma resultados previos en los que se registraban porcentajes de infección de 60 % para PVS (Vallejo et al., 2016), 66,6 % para PVY (Medina et al., 2015), 91,6 % para PLRV (Mesa et al., 2016) y 93,7 % para PVX (García et al., 2016).
En el mundo se han reportado más de 50 especies de virus en cultivos de papa (Kreuze et al., 2020), y aunque su efecto sobre los rendimientos es variable, pues depende de muchos factores socioeconómicos, tecnológicos y agroambientales, se ha estimado que las enfermedades virales pueden generar pérdidas del 50 % o aún más, con respecto al potencial productivo de las diferentes variedades de papa (Harahagazwe et al., 2018). En este sentido, Guzmán et al. (2012) encontraron que la infección por PYVV en papa criolla en Colombia provocaba reducciones en los rendimientos del 33 al 48 %. Del total de virus encontrados en papa, un importante número sólo se ha detectado en los andes suramericanos, incluyendo diferentes especies de los géneros Nepovirus, Tymovirus, Ilarvirus, Crinivirus, Cheravirus y Tepovirus (Kreuze et al., 2020); lo que refleja el hecho que esta región corresponde al centro de origen de este cultivo. Recientemente Fuentes et al. (2019) utilizando secuenciación de alto rendimiento (HTS) reportaron un gran número de nuevos virus en cultivos de papá en Perú, pertenecientes al menos a 14 géneros, incluyendo Badnavirus, Ophiovirus, Emaravirus, Comovirus, Potexvirus, Nucleorhabdovirus, Tobravirus y Nepovirus; siendo los virus asociados a este último género, algunos de los más numerosos. Hasta el momento en papa se han reportado seis especies de nepovirus: Cherry leaf roll virus (CLRV), Lucerne Australian latent virus (LALV), Tomato black ring virus (TBRV), Potato black ringspot virus (PBRSV), Potato virus U (PVU) y Potato virus B (PVb) (Kreuze et al., 2020). PBRSV, PVU y PVB sólo se han encontrado sobre papa en los Andes y su infección se ha asociado con síntomas del tipo calico y con diversos amarillamientos foliares (Kreuze et al., 2020).
El género Nepovirus hace parte de la familia Secoviridae (orden Picornavirales) y contiene 40 especies reconocidas de virus. Sus viriones consisten en partículas icosaédricas de cerca de 30 nm de diámetro con un genoma bipartita de ARN de cadena sencilla positiva (ARNss+) covalentemente unido en sus extremos 5' a una proteína VPg (genome-linked protein) y una cola de poli-A en su extremo 3' (Fuchs et al., 2017; Thompson et al., 2017). El ARN1 (7,2 - 8,4 kb) codifica para una poliproteína que es procesada por una proteasa viral en seis proteínas funcionales, incluyendo la polimerasa de ARN dependiente de ARN (RdRp) y la helicasa viral (NTB-protein); mientras que el ARN2 (3,7 7,3 kb) codifica para una poliproteína que una vez clivada da origen a la cápside (CP), la proteína de movimiento (MP) y la proteína P2A, involucrada en la replicación de este segmento genómico (Gaire et al., 1999; Fuchs et al., 2017). Filogenéticamente, los nepovirus se dividen en tres subgrupos (A, B y C), los que además difieren en la extensión del ARN2 y en los sitios de clivaje reconocidos por las proteasas virales (Fuchs et al., 2017).
Los nepovirus presentan rangos de hospedantes variables según la especie; en general inducen como síntomas principales manchas anulares, moteados y eventualmente amarillamientos. Se ha confirmado que al menos 12 especies de nepovirus son transmitidos de manera no persistente por nematodos longidóridos (Xiphinema, Longidorus o Paralongidorus spp.) y es frecuente su transmisión por polen y semilla sexual (Thompson et al., 2017). En las infecciones de nepovirus sobre plantas herbáceas es común que se presente el fenómeno de "recuperación de síntomas", lo que se ha asociado con la inducción del sistema de defensa antiviral de silenciamiento de ARN en sus hospedantes (Ghoshal y Sanfacon, 2015).
Con el fin de aportar nueva información al conocimiento del viroma de la papa criolla en Colombia, en este estudio se utilizó secuenciación de alto rendimiento (HTS) y RT-PCR cuantitativo (RT-qPCR) para la detección y caracterización molecular del Potato virus B (PVB), a partir de muestras de tejido foliar de plantas procedentes del oriente de Antioquia.
MATERIALES Y MÉTODOS
Muestras y extracción de ácidos nucleicos
Durante los meses de junio a septiembre de 2019, en 20 lotes de cultivo de S. phureja y 20 lotes de S. tuberosum (var. Diacol-Capiro) ubicados en el oriente de Antioquia (Colombia), se obtuvo una muestra compuesta formada por diez folíolos del tercio superior de plantas en estado de floración. Las muestras fueron obtenidas de manera aleatoria en un recorrido en zig-zag en las plantas ubicadas cada diez pasos al interior de los lotes de cultivo, evitándose así los efectos de borde. El ARN total fue extraído a partir de cada una de las muestras con el reactivo Trizol (Ambion - Thermo Fisher Scientific, Waltham, MA) a partir de 100 mg de tejido, siguiendo las instrucciones del fabricante. Paralelamente, se preparó una mezcla (bulk) de las muestras individuales de cada especie de papa y se realizó una extracción de ARN de doble cadena (ARNdc) a partir de 5 g de tejido siguiendo el protocolo de Valverde et al. (1990), pero con columnas de celulosa de fibra media (C6288 Medium) (Merck KGaA, Darmstadt, Alemania) y una elución final en 200 µL. Las colecciones se realizaron bajo el Permiso marco de recolección de especímenes biológicos otorgado por ANLA a la Universidad Nacional de Colombia (Resoluciones 0255 de 2014, 701 de 2016 y 1435 de 2018).
La concentración y pureza del ARN fue determinado por lecturas a 260 nm y 280 nm en un equipo Nanodrop 2000C (Thermo Fisher Scientific). La presencia de bandas de ARNdc se visualizó por corrido en gel de agarosa al 1,5 % teñido con GelRed (Biotium, San Francisco, CA) y una vez confirmada su presencia, se procedió a desnaturalizar 20 jjL a 98 °C por 5 min.
Secuenciación de alto rendimiento (HTS)
El ARNdc obtenido para las dos muestras (bulks) fue utilizado como base para la síntesis del ADNc con el kit TruSeq RNA Sample Preparation (Illumina, San Diego, CA), previa eliminación del ARN ribosomal (ARNr) con el kit Ribo-Zero (Illumina). En la generación de las librerías de ADNc se utilizó para la primera cadena la transcriptasa reversa SuperScript II (Thermo Fisher Scientific) y primers hexámeros en reacciones de 42 °C por 50 min; mientras que para la segunda cadena se empleó el SSM (Second Strand Master Mix) (Illumina) a 16 °C por 1 h y se siguieron las instrucciones del fabricante. La calidad y cantidad del ARN desnaturalizado y el ADNc sintetizado fueron evaluadas por el método del RIN (RNA Integrity Number) en un equipo 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Las muestras fueron enviadas a la compañía Macrogen (Corea del Sur) para ser secuenciadas mediante HTS en la plataforma Illumina NovaSeq.
Una vez obtenidas las secuencias, se realizó la remoción de las bases de baja calidad (Phred < 30) con el programa Sickle (Joshi y Fass, 2011). Luego se identificaron las secuencias del hospedante con BLASTN (Altschul et al., 1990) con un valor E (E-value) de 10-5 contra una base de datos del genoma de Solanum tuberosum (SolTub_3.0, GCF_000226075.1), Solanum lycopersicum (SPENNV200, GCF_001406875.1) y todos los transcritos de referencia disponibles en NCBI (https://www.ncbi.nlm.nih.gov/) para las plantas de la familia Solanaceae.
Posteriormente, las secuencias virales fueron identificadas con el programa Megablast (E-value: 10-5, qcov_hsp_perc: 90) contra una base de datos que incluye todas las secuencias de virus de plantas disponibles en la herramienta NCBI viral genomes resource (Brister et al., 2015). Los genomas de los virus identificados fueron ensamblados con la ayuda de genomas de referencia con el programa Magic-BLAST (Boratyn et al., 2019). Los genomas fueron confirmados mediante ensamblaje de novo con Trinity (Grabherr et al., 2011) y los contigs resultantes fueron evaluados por BLASTX contra una base datos de proteínas de virus de plantas (E-value: 10-5). Los ensamblajes fueron verificados con el paquete Integrative Genomics Viewer (IGV) (Robinson et al., 2011) y las secuencias consenso fueron anotadas con ayuda de la herramienta ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/) y BLASTX (Gish y States, 1993). Los análisis filogenéticos fueron realizados por el método Neighbor-Joining (Saitou y Nei, 1987) con las secuencias de aminoácidos de cada poliproteína con mil réplicas de bootstrap con el programa MEGAX (Kumar et al., 2018). El alineamiento de secuencias fue realizado con MUSCLE y las distancias calculadas con la matrix JTT y una distribución gama (Jones et al., 1992).
Detección de PVB por RT-PCR
A partir de cada una de las extracciones de ARN obtenidas para las 20 muestras individuales de ambas especies de papa, se realizó la síntesis de ADNc en 20 µL con 200 U de la enzima RevertAid Transcriptasa Reversa, 1X del buffer de RT, 0,5 mM de dNTPs, 20 U del inhibidor de ARNasas RiboLock (Thermo Fisher Scientific), 20 pmol del primer Oligo-dT y 3 µL de ARN (100-200 ng/µL), incubándose a 42 °C por 60 min y 70 °C por 10 min. El ADNc (50-100 ng) fue luego utilizado como molde para realizar reacciones de PCR cuantitativo (qPCR) con 0,3 µM de los primers Nepo_pol_F y Nepo_qpol_R diseñados en este estudio a partir de una región de RdRp del genoma del nepovirus PVB obtenido (Fig. 1) y el kit Maxima SYBR Green/ ROX (Thermo Fisher Scientific). El programa consistió en 10 min a 95 °C para activar la Taq polimerasa, seguido de 35 ciclos a 95 °C por 15 s y 52 °C por 60 s, en un equipo Rotor-Gene Q-5plex (Qiagen, Hilden, Alemania). Una vez finalizada la qPCR se determinaron los valores de ciclo umbral (Ct) y temperatura de fusión (Tm) en el rango 50-99 °C. En todas las reacciones se incluyó un control positivo y un control negativo, a partir de las primeras plantas de papa criolla identificadas en el laboratorio como infectadas o libres de PVB. Tres de los amplicones de la qPCR que resultaron positivos para el PVB fueron corridos en un gel de agarosa al 2 % y purificados directamente del gel con el kit GeneJET Gel Extraction (Thermo Fisher Scientific) para su posterior secuenciación Sanger en ambos sentidos en la compañía Macrogen (Corea del Sur).
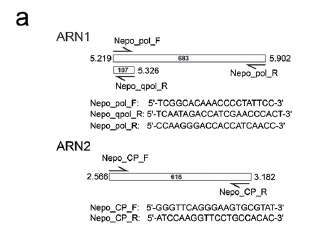
Figura 1 (a) Ubicación y secuencia de los primers diseñados para la detección del PVB mediante RT-qPCR y RT-PCR convencional.
En dos de las muestras que resultaron positivas para PVB mediante qPCR, se realizó también la amplificación por RT-PCR convencional de una región de la RdRp (684 pb) y la CP (617 pb) con los primers Nepo_pol_F/Nepo_pol_R y Nepo_CP_F/Nepo_CP_R diseñados en este estudio (Fig. 1) y siguiendo las condiciones utilizadas por Riascos Chica et al. (2018). Los amplicones fueron purificados directamente del gel, secuenciados en ambas direcciones y depositadas en GenBank con los códigos de accesión MT580275 y MT580276.
Uno de los fragmentos obtenidos por RT-PCR para la RdRp se clonó con el kit CloneJET PCR Cloning (Thermo Fisher Scientific) siguiendo las instrucciones del fabricante y se transformó en células competentes DH5a de Escherichia coli por choque térmico (Sambrook y Rusell, 2001). Las bacterias recombinantes se crecieron en cultivos con medio LB suplementado con ampicilina (50 mg/ml) y se extrajeron los plásmidos con el kit Plasmid Miniprep (Qiagen). La concentración de plásmidos fue determinada en un equipo Nanodrop 2000C (Thermo Fisher Scientific) y 0,22 µg fueron linearizados con la enzima XbaI (Thermo Fisher Scientific), para proceder a su transcripción in vitro con el kit TranscriptAid T7 High Yield Transcription (Thermo Fisher Scientific). El ARN obtenido se purificó por el método del fenol:cloroformo y su posterior precipitación con etanol absoluto (Sambrook y Rusell, 2001), cuantificándose nuevamente en Nanodrop 2000C (Thermo Fisher Scientific). Se usaron 1000 ng/µL del ARN transcrito como molde para la síntesis de ADNc con el cebador Nepo_qpol_R y 200 U de RevertAid H Minus reversa transcriptasa (Thermo Fisher Scientific), como se describió anteriormente. Finalmente, se prepararon diluciones seriadas de 10-1 hasta 10-7, a partir de una concentración inicial de 1000 ng/µl de ADNc, equivalente a 1,69 x 1013 copias virales/µl, de acuerdo con la fórmula:
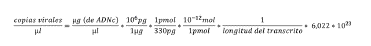
De esta forma, se procedieron a realizar las reacciones de qPCR por triplicado para cada una de las diluciones de ADNc y se construyó la curva estándar por comparación entre los valores de Ct para cada dilución y la concentración estimada de copias virales, con el software del equipo Rotor-Gene Q-5plex (Qiagen).
RESULTADOS
Secuenciación de alto rendimiento (HTS)
En la secuenciación HTS del ARN obtenido de la mezcla de muestras foliares de S. tuberosum, se obtuvieron 13 523 557 reads pareados y un total de 2 731 758 514 nt. Los análisis bioinformáticos identificaron que el 0,17 % de los reads (46 198) correspondían a secuencias asociadas a virus de plantas, siendo PVY el que presentó mayor profundidad de secuenciación (45 396 reads), aunque también se identificó el PMTV (ARN1: 54, ARN2: 42 y ARN3: 33 reads) (Fig. 2a). Para el transcriptoma de S. phureja se obtuvieron 21 692 655 reads pareados y un total de 4 381 916 310 nt. El 0,92 % de los reads (398 711) estuvo asociado a secuencias de virus de plantas; PVS (191 433 reads), PVV (150 004 reads) y PVX (23 381 reads) fueron los virus que presentaron mayor profundidad de secuenciación; también se identificaron secuencias de los virus: PVY (3641 reads), PVB (ARN1: 2166, ARN2: 1294 reads), BPEV (1710 reads) y PYVV (ARN1: 174, ARN2: 83, ARN3: 73 reads) (Fig. 2).
El hallazgo del PVB en cultivos de papa en Colombia es el primer registro de este virus por fuera del Perú (De Souza et al., 2017); los virus restantes ya han sido reportados en múltiples ocasiones para ambos hospedantes en esta región del oriente de Antioquia (Gutiérrez et al., 2016; Vallejo et al., 2016; Gallo et al., 2019; Sierra et al., 2020) y por tanto los resultados que se describen a continuación se centrarán en la caracterización del genoma de este nepovirus, cuya secuencia fue depositada en GenBank con los números de accesión MT521733 y MT521734 con el nombre del aislamiento PVB_phureja.
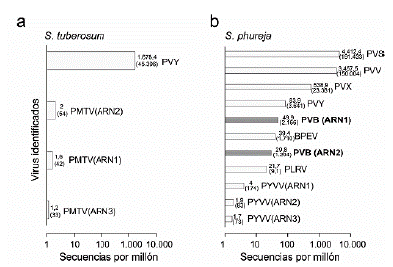
Figura 2 Virus identificados en los sets de datos obtenidos por HTS a partir de muestras de tejido foliar de (a) S. tuberosum y (a) S. phureja procedentes de cultivos en el oriente de Antioquia (Colombia). Entre paréntesis se presenta la totalidad de reads obtenidos para cada genoma viral. En negrita se resalta la identificación de reads asociados con los dos segmentos genómicos del PVB.
El ARN1 de este virus presentó 7126 nt, excluyendo la cola de poli-A, que corresponden al 99,7 % de la secuencia de PVB_Pasco-01 (NC_043447) reportada por De Souza et al. (2017). La secuencia obtenida carece de los primeros veinte nucleótidos. Las regiones no traducidas en los extremos 5' y 3' tienen longitudes de 120 y 211 nt (sin poli-A), respectivamente. En la región UTR 3' se identificó un motivo de estructura secundaria también presente en el UTR 3' del ARN2 (295 nt). El ARN1 presenta un único ORF que codifica para una poliproteína (P1) de 2264 aminoácidos (aa), ubicado entre las posiciones 121 y 6912, codón de inicio AUG con una secuencia Kozak AatATGc, donde la citosina en posición +4 difiere de la G esperada (Kozak, 2005) y un codón de finalización UGA (Fig. 3). La búsqueda de motivos conservados en este segmento genómico indicó la presencia de regiones con motivos típicos de un cofactor de proteasa (Pro-cof), una helicasa dependiente de NTB (Hel), una cisteín-proteasa (Pro), una proteína de unión al genoma (VPg) y una polimerasa de ARN dependiente de ARN (RdRP); hasta el momento se desconoce la función del primer polipéptido (X1) de este segmento genómico (Fuchs et al., 2017; Thompson et al., 2017) (Fig. 3). La herramienta prosite (https://prosite.expasy.org/) detectó un motivo característico de cisteín-proteasas 3c/3c-like de picornavirales (PS51874, PCV_3C_PRO, score= 43,712) en las posiciones 12361445. Para la región helicasa se detectaron los tres motivos funcionales característicos de la superfamilia 3 de este tipo de proteínas de virus de ARN de cadena sencilla positiva: A (GKRHCGKS), B (D) y C (N), ubicados en las posiciones 792-799, 842 y 889, respectivamente (Gorbalenya y Koonin, 1993), lo que fue confirmado mediante la herramienta Pfam (Finn et al., 2014) que detectó el dominio ARN helicasa (PF00910, 3,7e-23) entre las posiciones 788-890.
Más adelante se encontró el motivo DRGYRARNU.PVNHR característico de proteínas VPg en la subfamilia Comovirinae en las posiciones 1213-1239 (Mayoa y Fritsch, 1994). En el extremo C-terminal de la poliproteína se identificó la triada catalítica H, E/D, y C altamente conservada en cisteín-proteasas y finalmente la RdRp se extiende entre los residuos 1450-2264; la herramienta Pfam (Finn et al., 2014) detectó el dominio de ARN polimerasa dependiente de ARN (PF00680, E-value 3,9e-77) entre las posiciones 1478-1964 que además contiene el motivo DX4DX59GX3TX3NX33GDD típico de estas proteínas en la región 1727-1836 (Argos, 1988; Koonin y Dolja, 1993). Los sitios putativos de clivaje de esta poliproteína previamente reportados para PVB (De Souza et al., 2017) fueron R427/A428, R617/S618, K1212/ S1213, R1239/S1240, K1449/S1450 (Fig. 2).
El ARN2 de este virus presentó 4298 nt, excluyendo la cola de poli-A que corresponde al 94,9 % respecto a la secuencia de referencia del PVB_Pasco-01. Para el extremo 5' no se obtuvieron los primeros 227 nt, incluyendo los primeros cinco codones del ORF que codifica para la poliproteína P2 (1371 aa) que comprende las posiciones 1- 4100 nt. El extremo 3' consistió de 295 nt (sin poli-A). Una vez es procesada P2 por la cisteín-proteasa viral, se generan las proteínas putativas 2A (24 kDa), la proteína de movimiento (MP) y la proteína de la cápside (CP). El motivo conservado P identificado por Mushegian (1994) en la MP de los nepovirus se ubica en la posición 627. La proteína 2A corresponde a la región N-terminal; se cree que esta proteína es requerida para la replicación del ARN2 y se co-localiza con los complejos de replicación viral (VRCs) que contiene tanto proteínas virales como factores del hospedante (Gaire et al., 1999; Fuchs et al., 2017). Con base en comparaciones realizadas con otros nepovirus, De Souza et al. (2017) identificaron el sitio putativo de clivaje para separar MP de CP como L812/K813, que para el aislamiento PVB_phureja se identificó en la misma posición, con el mismo sitio de corte (Fig. 3).
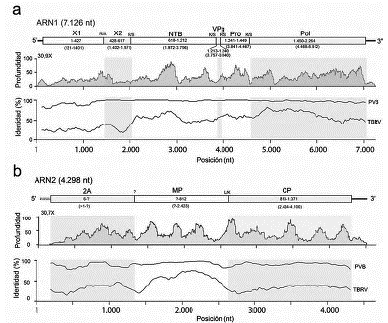
Figura 3 Profundidad de secuenciación de los dos segmentos genómicos (ARN1 y ARN2) del aislamiento PVB_phureja obtenido en muestras de S. phureja del oriente de Antioquia (Colombia) por HTS. En la parte inferior se indican los niveles de identidad de nucleótidos del genoma de PVB_phureja con respecto a la cepa PVB_ Pasco-01 obtenida en Perú y de otro nepovirus del subgrupo B (TBRV). En la barra superior se presentan los diagramas de los dos segmentos genómicos de PVB_phureja, indicándose la posición de las regiones codificantes para sus diferentes proteínas y los sitios de corte putativos de las proteasas virales. Las posiciones de aminoácidos para cada proteína putativa se indican al interior de las barras, mientras que las posiciones de los nucleótidos respectivos se marcan entre paréntesis en la parte inferior de las barras.
Según el ICTV, el criterio para la separación de especies en la familia Secoviridae corresponde a virus con niveles de identidad en la secuencia de aa inferiores al 80 % para la región comprendida entre los motivos CG y GDD de la proteasa y la RdRp, respectivamente (región Pro/Pol) e inferior al 75 % para CP. Adicionalmente, diferentes propiedades biológicas como rango de hospedantes, vector específico, ocurrencia de rearreglos entre los dos segmentos de ARN, ausencia de protección cruzada y diferencias en las reacciones antigénicas, son otros criterios que se tienen en cuenta para la definición de especies en el género Nepovirus (Thompson et al., 2017). Las comparaciones realizadas entre las regiones Pro/Pol y CP del aislamiento PVB_ phureja y las secuencias de aminoácidos del aislamiento Pasco-01, indicaron porcentajes de identidad de 98,1 % y 90 %, respectivamente, lo que confirma su pertenencia a dicha especie. Estos niveles de identidad cayeron a valores inferiores al 71,5 % con respecto a otros nepovirus del subgrupo B (Tabla 1). A nivel de la secuencia de aminoácidos de las poliproteínas completas, los niveles de identidad del PVB_phureja con respecto al PVB_Pasco-01 fueron del 96,1 % y 90 % para P1 y P2, respectivamente. Los análisis fi logenéticos realizados con base en la secuencia de aminoácidos de cada poliproteína (P1 y P2), ubicaron al aislamiento PVB_phureja en el clado correspondiente al subgrupo B de los nepovirus, en conjunto con GCMV, GARSV, BRSV y TBRV, entre otros (Fig. 4)
Tabla 1 Niveles de identidad de aminoácidos y nucleótidos para la región Pro/Pol ubicada entre los motivos CG de la proteasa y GDD de RdRp en la poliproteína 1 y para la CP de la poliproteína 2 del aislamiento PVB_phureja en comparación con diferentes especies de nepovirus, incluyendo el aislamiento de referencia PVB_Pasco-01 del Perú.


Figura 4 Árboles filogenéticos generados por el método Neighbor-joining basados en la secuencia de aminoácidos de las poliproteínas codificadas por el ARN1 y ARN2 de diferentes nepovirus. En negrita se resalta el aislamiento PVB_phureja obtenido en el presente estudio. Los números sobre las ramas indican los valores de bootstrap.
Detección de PVB por RT-PCR
Con base en la secuencia de la RdRP del ARN1 del genoma del PVB_phureja obtenida en este estudio, se diseñaron primers para la detección específica de este virus por medio de RT-qPCR. Los resultados indicaron la utilidad de los primers, lográndose la detección del PVB en siete de las 20 muestras (35 %) de tejido foliar de S. phureja evaluadas, mientras que ninguna de las 20 muestras de S. tuberosum resultó positiva para este virus (Fig. 5c). Las secuencias de tres de los amplicones de 107 pb obtenidos por RT-qPCR se confirmaron por secuenciación Sanger como pertenecientes a la región RdRp de PVB_phureja, mientras que dichos niveles fueron inferiores al 90 % con respecto a otros nepovirus cuyas secuencias se encuentran disponibles en GenBank (Artichoke Italian latent virus [AILV-V]: NC_043684, Beet ringspot virus [BRSV]: MF141079, Cycas necrotic stunt virus [CNSV]: MK512741). Los valores de ciclo umbral (Ct) obtenidos para las muestras se ubicaron en el rango de 21,9 a 33,57, mientras que con la herramienta HRM (High Resolution Melting) se encontró un valor de Tm de 79 ± 1 °C (Fig. 5d).
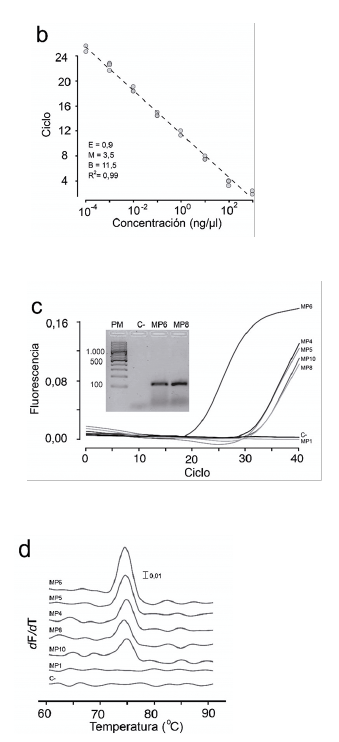
Figura 5 (b) Curva estándar realizada para la cuantificación absoluta del PVB mediante RT-qPCR. (c) Curvas de amplificación de RT-qPCR con SYBR Green para una región de 107 pb de la región RdRp, y la confirmación respectiva en gel de electroforesis al 2 % de los amplicones obtenidos. (d) Curvas de fusión para diferentes amplicones generados por RT-qPCR con un valor de Tm de 79 ± 1 °C para las muestras positivas para PVB.
La cuantificación absoluta del número de copias virales presente en cada muestra con la ecuación de la curva estándar obtenida en el estudio (y = -3,496x + 11,546, donde Y=Ct yx= Log del número de copias virales/^L) indicó la ocurrencia de una carga viral en el rango de 9x1010 -1,06x1011 copias virales/(jL (Fig. 5b).
La secuenciación Sanger de los amplicones obtenidos para la RdRp y la CP, indico niveles de identidad del 87,2 % y 86,2 % con las secuencias de PVB disponibles en GenBank (i.e. NC_043447, NC_043448), respectivamente.
La identidad de las secuencias Sanger con relación a la región respectiva en el genoma consenso obtenida por HTS del aislamiento PVB_phureja fue del 100 % (RdRp) y 99,8 % (CP). En la figura 3 se presenta una comparación de los niveles de identidad local (ventana 100 nt) de PVB_ phureja con respecto a la secuencia de PVB reportada en Perú por De Souza et al. (2017) y para TBRV. De estas comparaciones se destaca el hecho que se presentaron 899 y 548 nt polimórficos para el ARN1 y para el ARN2 de los dos aislamientos de PVB evaluados, además de la ocurrencia de 87 y 136 cambios en aa para la P1 y P2, respectivamente, lo que indica que este virus presenta muy altos niveles de variación intraespecífica.
DISCUSIÓN
En este estudio mediante HTS y confirmación por RT-qPCR se detectó por primera vez en Colombia el nepovirus PVB, un virus de ARNss+ con genoma bipartita asociado en otros estudios con síntomas tipo calico y que hasta ahora sólo había sido reportado en Perú (De Souza et al., 2017; Fuentes et al., 2019). Recientemente, se encontró que este virus tiene una amplia distribución geográfica en ese país suramericano, siendo detectado mediante HTS en al menos 195 muestras de siete departamentos diferentes y en un amplio rango de variedades nativas y/o mejoradas de papa como Huayro, Yungay y Canchán (Fuentes et al., 2019). Análisis filogenéticos realizados por De Souza et al. (2017) habían demostrado la ubicación de esta especie en el subgrupo B del género Nepovirus, siendo claramente diferenciado de las otras cinco especies de nepovirus hasta ahora reportadas en papa: PBRSV, PVU, CLRV, LALV, TBRV (Kreuze et al., 2020). El aislamiento PVB_phureja aquí secuenciado a partir de tejido foliar asintomático de papa criolla en Antioquia, compartió un 87,4 % y 86,9 % con el aislamiento PVB_Pasco-01 de Perú para la secuencia de nucleótidos del ARN1 y ARN2, respectivamente; mientras que dichos valores fueron de 98,1 % y 90 % para la secuencia de aminoácidos de la región comprendida entre los motivos CG de Pro y GDD de Pol y para la CP, respectivamente. Esto confirma su identidad como PVB, pues entre los criterios definidos por el ICTV para la delimitación de especies en la familia Secoviridae se destacan la ocurrencia de niveles de identidad de aminoácidos menores a 75 % y 80 % para CP y Pro/Pol, respectivamente (Thompson et al., 2017).
La ocurrencia de altos niveles de variabilidad entre aislamientos de una misma especie es una característica muy común en los nepovirus, lo que se cree es el resultado de la alta frecuencia de procesos de rearreglo de los dos segmentos genómicos entre cepas, y/o de la recombinación intra e interespecífica que ocurre en estos virus. Por ejemplo, el ARN2 de diferentes aislamientos de GFLV puede presentar niveles de identidad del 78,3 % nt, mientras que la secuencia de aminoácidos de la poliproteína (P2) de hasta 86,3 % (Fuchs et al., 2017). Una situación similar fue reportada por De Souza et al. (2017) para PVB, al encontrar con base en secuencias de RdRp, dos grupos de aislamientos (G1 y G2) que presentaban niveles de identidad en la secuencia de nucleótidos del 86,8 %, mientras que al interior de cada grupo este valor fue de 98,7 % y 95,7 %, respectivamente. El aislamiento PVB_phureja obtenido en este estudio está relacionado con el Grupo 2 de PVB, dados los altos niveles de identidad que presenta con los aislamientos de este grupo.
Por otra parte, las comparaciones realizadas para la secuencia de aminoácidos de las regiones Pro/Pol y CP de PVB_phureja con respecto a otros nepovirus, indicaron que las especies TBRV y BRSV fueron las más cercanas con niveles de identidad de 71,5 % y 68,8 % para Pro/Pol; mientras que para la región CP estos niveles fueron del 42 % y 41,6 % con respecto a TBRV y GCMV, respectivamente. Estos valores son muy cercanos a los reportados para el PVB_Pasco-01 con respecto a otros nepovirus del subgrupo B, en los que se reportaron valores de identidad del 53-71 % para Pro/Pol y del 25-34 % para CP (De Souza et al., 2017).
Con el fin de evaluar la prevalencia del PVB en cultivos de papa en Antioquia, se diseñaron un par de primers específicos con base en la región que codifica para RdRp para su utilización en pruebas de RT-qPCR. Este virus fue detectado en 7 de las 20 muestras de papa criolla, pero en ninguna de las 20 muestras de S. tubersoum var. Diacol-Capiro. Ya que el PVB se ha reportado en Perú sobre diferentes variedades de S. tuberosum (Fuentes et al., 2019), es necesario continuar con la evaluación del virus en otras regiones de cultivo y variedades de esta especie de papa en Colombia, antes de descartarla como su hospedante en el país.
El rango de Ct para las muestras que resultaron positivas para PVB fue muy variable (21,9 a 33,6), con un estimado de copias virales desde 9,02x1010 a 1,06x1011, obtenidas mediante cuantificación absoluta con la curva estándar generada en este estudio. Cuando se realizó la evaluación de Tm, se encontró que esta se presentaba en el rango de 79 ± 1 °C, lo que indica la presencia de variaciones en las secuencias de los aislamientos del virus, aún para esta pequeña región del genoma (107 pb). Así mismo, la utilidad de los primers diseñados (Nepo_pol_F/Nepo_pol_R y Nepo_CP_F/Nepo_CP_R) para su utilización en RT-PCR convencional fue validada por secuenciación Sanger para ambas regiones, obteniéndose un 100 % de identidad para el amplicon obtenido para RdRp y del 99,8 % para la región parcial de CP con respecto al ensamblaje conseguido por HTS.
La detección del PVB en el 35 % de las 20 muestras obtenidas en igual número de lotes de papa criolla del Oriente de Antioquia resulta preocupante para el gremio cultivador de esta especie de papa en Colombia, y en conjunto con resultados previos que reportan niveles de prevalencia superiores al 40 % de virus como PVY, PLRV, PVS, PVX y PVV (Medina et al., 2015; García et al., 2016; Mesa et al., 2016; Vallejo et al., 2016; Sierra et al., 2020) en tubérculos-semilla comercializados en Antioquia, deben llamar la atención de los organismos de sanidad vegetal del país sobre la necesidad de fortalecer el programa de certificación de semilla de S. phureja.
La detección del PVB en cultivos de papa criolla en Colombia representa el primer reporte de este virus por fuera de Perú; el genoma reportado para el aislamiento PVB_ phureja, así como los primers diseñados para su detección en pruebas de RT-qPCR y RT-PCR convencional permitirán abordar diferentes preguntas biológicas sobre este virus, que incluyen su rango de hospedantes, posible transmisión por nematodos longidóridos, síntomas específicos que induce en papa criolla y sus efectos sobre el rendimiento o la calidad de esta especie de papa en Colombia y otros países andinos.
CONCLUSIONES
Mediante secuenciación HTS del transcriptoma de tejido foliar de plantas de S. phureja obtenidas en el Oriente de Antioquia se detectó por primera vez en Colombia el nepovirus PVB, siendo posible secuenciar la mayor parte de sus dos segmentos genómicos de ARNss+ (ARN1: 7126 nt, ARN2: 4298 nt), los que codifican para las poliproteínas P1 de 2264 aa y P2 de 1371 aa, respectivamente. Análisis filogenéticos realizados para las dos poliproteínas (P1 y P2) ubicaron al aislamiento PVB_phureja en el subgrupo B de los nepovirus, en conjunto con el TBRV, BRSV, GCMV, GARSV, AILV y con el aislamiento PVB_Pasco-01 de Perú, con quien compartió niveles de identidad de 98,1 % y 90 % con P1 y P2, respectivamente. Se diseñaron primers específicos para la región RdRP útiles para la detección del PVB en pruebas de RT-qPCR con SYBR Green, encontrándose este virus en el 35 % de las 20 muestras de papa criolla evaluadas y en ninguna de las 20 muestras de papa var. Diacol-Capira. Para esta prueba se diseñó una curva estándar que permitió cuantificar la carga viral del PVB en las muestras positivas en un rango de 9,02x1010 a 1,06x10" copias virales/µL y con valores de Tm de 79 ± 1 °C.

