











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La encefalitis autoinmune es una de las causas más comunes de encefalitis aguda no infecciosa, inmunomediada en 20 a 41% de los casos según lo reportado en diferentes cohortes 1,2. El diagnóstico en la práctica clínica es un desafío puesto que los resultados de anticuerpos no están siempre disponibles, las pruebas negativas no excluyen el diagnóstico y el debut y la evolución clínicas son muy variadas 3. El diagnóstico se orienta a partir de las manifestaciones clínicas, la presencia de biomarcadores, los paraclínicos que descartan otras probables causas. En nuestros casos la presentación clínica se manifiesta con epilepsia, fluctuaciones de conciencia, cambios comportamentales y afectivos, movimientos anormales y alteración autonómica. Encuadrar esta variabilidad resulta fundamental para sospechar, diagnosticar y realizar estudios complementarios que lleven oportunamente a la confirmación y tratamiento dirigido y a continuar los estudios para el probable origen primario.
Caso 1
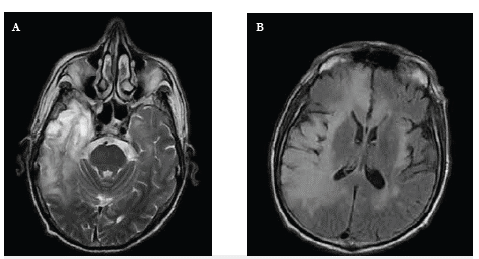
Un hombre de 64 años consultó por dos meses de vértigo e inestabilidad para la marcha, seguido de estado epiléptico focal y deterioro de la conciencia hasta llevarlo a estupor. La resonancia magnética (RM) cerebral mostró reacción inflamatoria en el lóbulo temporal derecho con extensión al lóbulo frontal (figura 1), y el o líquido cefalorraquídeo (LCR) hiperproteinorraquia y pleocitosis linfocitaria con PCR para virus Epstein Barr y herpes negativos. Por considerarse inicialmente de probable etiología viral, recibió manejo con aciclovir presentando ligera mejoría clínica. Posteriormente presentó fluctuaciones del estado de conciencia y compromiso conductual. Se realizó biopsia cerebral con áreas aisladas de infiltrado inflamatorio linfoide de patrón reactivo con células T CD3 y CD5, y células B CD20. Este hallazgo, más anticuerpos anti'NMDA (N- metil- D- aspartato) positivos detectados en LCR soportaron el diagnóstico de encefalitis límbica de origen autoinmune. Tras la administración de metilprednisolona con escasa mejoría, por lo que se inició inmunoglobulina G , recuperó el nivel de conciencia y resolvieron las alteraciones comportamentales y focales, sin secuelas.
Caso 2
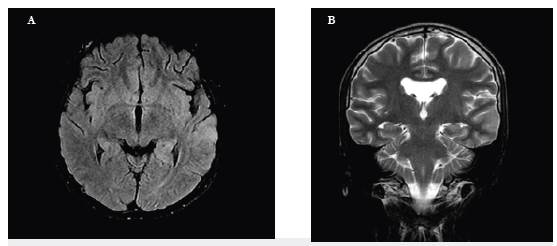
Un adolescente de 16 años debutó de forma aguda con insomnio, agresividad, ideación paranoide, alucinaciones visuales y auditivas, con LCR normal, enfocado como primer episodio psicótico agudo de difícil manejo. Fue tratado con risperidona (6mg en total), haloperidol (20mg totales), midazolam (20mg en 24 horas) y pipotiazina depósito 50 mg con posterior taquicardia, fiebre, leucocitosis, elevación de creatinina quinasa (1896 mg/ dl) y una convulsión tónico clónica generalizada. Si bien se consideró como síndrome neuroléptico maligno (SNM), la RM cerebral mostró hiperintensidad cortical en secuencia T2 temporal izquierda (figura 2), con anti NMDA, herpes virus, perfil autoinmune y paraneoplásico negativos. No respondió a aciclovir, con respuesta parcial a inmunoglobulina G (IGIV), con control de movimientos anormales pero con persistencia de compromiso mental. A los 10 días de inicio de los síntomas cursó con estado epiléptico super-refractario que resolvió tras la realización de recambios plasmáticos, con recuperación completa después de tres meses de rehabilitación.
Caso 3
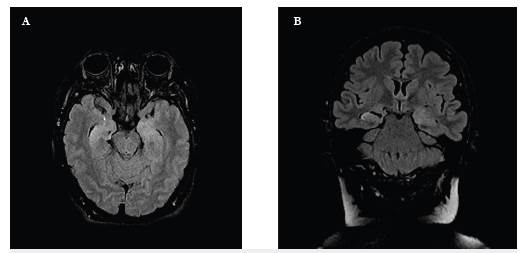
Una mujer de 45 años, con antecedente de adenoma de parótida benigno, cursó con cefalea, fallas en orientación espacial y de memoria a corto plazo, de un mes de evolución, seguido de apatía, llanto fácil e ideas de minusvalía. A los dos meses del inicio del cuadro presentó crisis focales que iniciaron con quejido, piloerección del hemicuerpo izquierdo indicando compromiso disautonómico, seguido de crisis tónico-clónica bilateral de difícil control configurando un estado epiléptico controlado con benzodiacepina y ácido valproico. Así mismo, presentó posturas distónicas facio-braquiales derechas sin correlación en EEG. En RM cerebral se observaron hiperintensidades T2 temporales mesiales bilaterales (figura 3) con PCR para virus del herpes simple (VHS) negativa. Pesquisa paraneoplásica negativa. Los síntomas mejoraron y las crisis cedieron tras la administración de 2g de inmunoglobulina en cinco días, con fallas secuelares en memoria de trabajo.
Caso 4
Un hombre de 84 años debutó con cefalea, alucinaciones visuales, pensamiento incoherente y pérdida del control de esfínteres. LCR con pleocitosis linfocitaria y RM con lesiones hiperintensas en T2 en regiones frontales mediales bilaterales, frontal basal izquierda e ínsulas bilaterales. Presentó deterioro con agitación psicomotora y rápido declive en lenguaje y memoria a pesar de tratamiento antiviral y antibiótico, con episodios de fluctuación del estado de conciencia, llegando al coma y relacionando mejoría al aplicarse bolos de corticoides y deterioro progresivo posteriormente. Dentro de los estudios se encontraron descargas de punta-onda lenta fronto-temporales izquierdas en EEG y con progresión imagenológica de lesiones. En búsqueda de una etiología de encefalitis posiblemente paraneoplásica o autoinmune se amplió pesquisa, encontrándose lesión plantar de la cual se tomó biopsia confirmándose sarcoma de Kaposi clásico maculonodular.
DISCUSIÓN
Definición
La encefalitis autoinmune tiene una incidencia estimada de 0,8 por 100.000 4 y se caracteriza por la reacción auto-inmune en el sistema nervioso central por acción de anticuerpos antineuronales (intracelulares o de superficie), que se correlacionan con manifestaciones clínicas especificas, incluyendo alteración del estado de conciencia, trastornos psiquiátricos, movimientos anormales, disautonomías, estatus epiléptico entre otros (tablas 1 y 2)
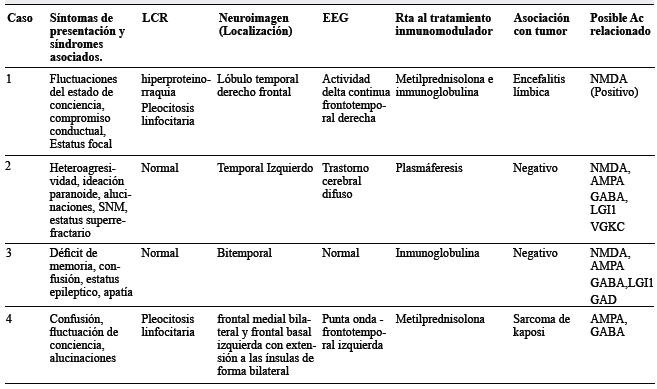
Tabla 1 Descripción de las características clínicas, las pruebas diagnósticas, tratamiento y posible Ac relacionado en nuestros pacientes con sospecha de Encefalitis Autoinmune.
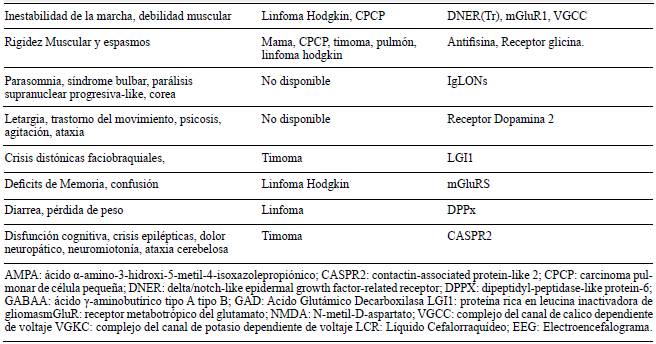
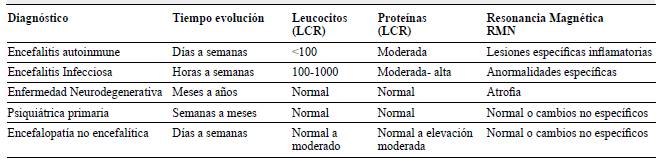
Dada la variabilidad clínica, en su inicio puede confundirse con múltiples causas, incluyendo inicio agudo de psicosis, infecciones del sistema nervioso, vasculitis, disautonomías primarias y secundarias, entre otras (tabla 3.)
En cuanto a su fisiopatología, si bien no está del todo dilucidada, se sabe que tiene una base multifactorial donde intervienen tanto factores genéticos como medioambientales 5. A lo largo de los años, se han caracterizado autoanticuerpos asociados con encefalitis autoinmune y en un amplio modo se han identificado dos tipos diferentes: contra dominios extracelulares de antígenos específicos de la neuroglia (antígenos de la superficie celular): AMPAR, CASPR2, DPPX, GABA, GlyαR, IgLON5, mGluRl, mGluR5, NMDAR, VGKC, VGCC y de ubicación extra-celular: LGI-1 6 y otros contra antígenos en las células neuronales: AGNA, ANNA, CRMP-5. GAD-65, GFAP, anti-Ma, PCA, que guardan estrecha relación con síndromes paraneoplásicos clásicos con una respuesta no muy adecuada frente a la inmunoterapia. 7,8
La característica que más resalta en los antígenos descritos en primer lugar es su rápida accesibilidad a todos los componentes del sistema inmune 6, facilitada a su vez por una alteración en la homeostasis de la barrera hematoencefálica (BHE). Si bien existe una producción intratecal de autoanticuerpos, se ha evidenciado el paso de estos a través de una BHE permeada, asociado además a infiltración no controlada de linfocitos B. Lo anterior se ha identificado a través de ensayos de metilación de ADN de todo el genoma que los genes S100A6 promueven la penetración del linfocito B a la capa endotelial de la BHE 9. En un estudio realizado en 68 pacientes con encefalitis autoinmune se evaluó el índice de CD4/CD8+ con citometría de flujo encontrando que una proporción más baja de células T CD4 / 8 + en el LCR se asocia con una disfunción de la BHE 10, y adicionalmente que moléculas inflamatorias como IL-1β, FNT-α, CCL-2 y la IL-17A juegan un papel importante en la disregulación inflamatoria generando disrupción de las uniones celulares, expresión de metaloproteasas y activación de receptores tipo Toll (TLRs) 11. Se ha demostrado un vínculo directo entre la infección y la encefalitis autoinmune, como es el caso de pacientes con encefalitis por VHS, en los que se puede presentar recurrencia de deterioro neurológico hasta un 10-25% y el tratamiento antiviral no es efectivo, sin embargo, el manejo inmunomodulador produce mejoría clínica. Hasta 64% de estos pacientes desarrollan anticuerpos NMDAR en suero o LCR, por lo que se debe considerar la posibilidad de encefalitis autoinmune postinfecciosa en pacientes con síntomas recurrentes, como el primer caso presentado, en quien se presentó leve mejoría clínica con el manejo con aciclovir con un posterior deterioro. 40
Otro aspecto a resaltar es el componente maligno subyacente causado por la respuesta inmune contra los antígenos neuronales expresados por los tumores 6 donde si bien los autoanticuerpos contra antígenos de superficie se relacionan más con patología idiopática, en casos como NMDA puede presentarse hasta un 50% de síndromes paraneoplásicos. En contraparte autoanticuerpos contra agentes intracelulares como: Hu, Ri, CV2/CRMP5, Ma2 son altamente sugestivos de un síndrome paraneoplásico, por lo que debe buscarse la malignidad subyacente, donde clásicamente se encuentra carcinoma pulmonar (de pequeñas células principalmente), tumores de células germinales y timomas 12
Manifestaciones clínicas
En cuanto a los síntomas psiquiátricos, suelen ser las principales manifestaciones en dos terceras partes de los pacientes con encefalitis autoinmune confirmada. Estos pacientes son usualmente evaluados y manejados en instituciones psiquiátricas sin recibir un diagnóstico y tratamiento dirigido para encefalitis autoinmune. Los síntomas incluyen cambios comportamentales agudos (56%), alucinaciones visuales y auditivas, confusión y agresión (18%), ideas paranoides (17%), depresión (13%), catatonia (10%), mutismo (8%) y anorexia (1%) 13. En un estudio con seguimiento a tres años después del tratamiento, 50% de los pacientes continuaban con síntomas psiquiátricos 14. Aunque esta presentación es muy sugestiva de encefalitis anti-NMDAR, también se puede presentar en anti-AMPAR y anti-GABA-B-R15. Retrospectivamente en 111 pacientes con diagnóstico de encefalitis autoinmune, 52 cursaron con SNM. Kiani y cols. presentaron dos pacientes con autismo y discapacidad intelectual, con encefalitis antiNMDA asociada a catatonia maligna, alteración de conciencia, inestabilidad autonómica, mutismo, rigidez y posturas anormales que se pueden confundir con SNM; ambas condiciones pueden cursar con leucocitosis y elevación de la CK 16 Esto sugiere una posible relación patofisiológica entre SNM, catatonia maligna y encefalitis autoinmune, y se propone una alteración en la señalización de los receptores neuronales NMDA, que modifica a su vez al receptor dopaminérgico por medio de interacción directa entre el receptor y la cascada de señalización intracelular 17. También se ha postulado una interacción entre la proteína LGI1 y el sistema glutaminérgico como factor común 18.
En nuestros casos la respuesta a la inmunoterapia orientó hacia una etiología autoinmune de la encefalitis, a pesar de que en 3 de 4 casos los anticuerpos antiNMDA fueron negativos. La presentación con síntomas neuropsiquiátricos como la característica principal sugiere la presencia de anticuerpos contra el complejo del canal de potasio dependiente de voltaje (VGKC) 19.
La epilepsia, en especial del lóbulo temporal, es otra manifestación que afecta hasta 26% acorde a una serie de 84 pacientes con epilepsia del lóbulo temporal de inicio tardío, en quienes se identificó encefalitis autoinmune en el 27%; de este último grupo 26% fue paraneoplásica 20. Los blancos neuronales más comunes son: el receptor NMDA, el receptor AMPA (α -amino-3-hidroxil-5-metil-4-isoxazol-propionato), el receptor GABA A y B (acido gamma amino butírico) y LGI1 (proteína rica en leucina inactivadora de gliomas) y antígenos neuronales intracelulares como el GAD (acido glutámico decarboxilasa) 21.
Los movimientos anormales tipo distonías faciobra-quiales, como en nuestro tercer caso, son característicos de encefalitis por LGI1; incluso pueden preceder la enfermedad meses antes 22, usualmente se asociano a deterioro cognitivo progresivo así como a síntomas disautonomicos 23 por afección de los sistemas dopaminérgico, adrenérgico y colinérgico, asociándose también a una peor respuesta y pronóstico 24. Si bien no fue el caso de nuestra paciente ante dichas caracteristicas clinicas se deberá contemplar la solicitud específica del anticuerpo según la manifestación. Para este grupo se prefiere el tratamiento específico de la neoplasia o terapia inmunosupresora. Como segunda línea se puede incluir rituximab y ciclofosfamida 25.
La relación causal entre encefalitis autoinmune y sarcoma de Kaposi causado por el herpes virus humano 8 (HVH8) no pudo ser comprobada en el cuarto caso, sin embargo se han descrito series de casos de metástasis intracraneales de sarcoma de Kaposi visceral 26, de leucoencefalopatía multifocal progresiva 27 y linfoma primario de sistema nervioso central asociados a HVH8. La relación entre el virus HVH8 y autoinmunidad no está completamente dilucidada pero en la enfermedad de Castelman 28 la autoinmunidad secundaria a HVH8 y la introducción de su ADN en las células blancas crea sobreexpresión de dicha proteína cuyo material para transcripción se encuentra en el ADN viral y al generar esta proteína, se comportaría como una citoquina activadora de linfocitos B, estimulando la producción de autoanticuerpos soportándose aún más la teoría autoinmune dada la mejoría con el manejo inmunomodulador 29.
De acuerdo con el algoritmo diagnóstico de Dalmau y Graus, todos nuestros pacientes cumplen con el diagnóstico clínico de encefalitis autoinmune posible 30, dado el inicio subagudo (progresión en menos de 3 meses), fluctuación del estado de conciencia, síntomas psiquiátricos, fallas de memoria, signos de focalización neurológica, convulsiones, en algunos con pleocitosis en LCR y el EEG con actividad epiléptica en los lóbulos temporales (evidenciado solo en uno de nuestro casos con actividad frontotemporal) junto con las anormalidades en resonancia en secuencias con información T2 altamente restringidas a los lóbulos temporales mesiales 30.
Los hallazgos en el LCR pueden ser anormales en 79% de los casos descritos en la literatura 31. Las características más frecuentes son: pleocitosis linfocitaria (90%), hiper-proteinorraquia (33%) y presencia de bandas oligloclonales (25%) 32, sin embargo, estos son hallazgos inespecíficos 33. El LCR puede encontrarse normal lo que no descarta la enfermedad y debe estudiase como una etiología probable 34. Respecto al EEG los patrones periódicos o rítmicos, las convulsiones y el estado epiléptico refractario de nueva aparición confieren un mayor riesgo de mal resultado, independientemente del subtipo de encefalitis autoinmune. 35
Para realizar un diagnóstico definitivo es necesaria la detección de anticuerpos contra proteínas sinápticas, de superficie celular u onconeurales, sin embargo, en un análisis retrospectivo multicéntrico en el que se aplicaron pruebas para detección de anticuerpos neuronales en 118 pacientes que cumplían criterios clínicos para encefalitis autoinmune se concluyó que aquellos con anticuerpos positivos y negativos mostraban un perfil similar en término de frecuencia de tumor. Los pacientes con anticuerpos negativos fueron el fenotipo más común; la manifestación clínica más frecuente fueron crisis; 60% respondieron a la terapia, sin embargo la ausencia de anticuerpos positivos implicó un retraso en el inicio de la terapia de primera línea, lo que se asociaba a pobre respuesta y peor pronóstico 36. Este hallazgo se soporta en reportes que muestran que 44% de los pacientes que responden a la terapia inmunosupresora tienen encefalitis autoinmune con anticuerpos no detectables 30.
TRATAMIENTO
Los corticosteroides, la IGIV o la plasmaféresis son opciones de primera línea, y en caso de falla, pero con alta sospecha o con componente inmune comprobado puede requerirse una terapia de segunda línea, como rituximab, ciclofosfamida o micofenolato, así como en aquellos casos refractarios o que tienen recaídas recurrentes 37. El uso temprano del tratamiento de inmunoterapia impacta en los desenlaces, por lo que no debe retrasarse su inicio con tasas de éxito cercanas a 80%, apoyando así la impresión diagnóstica de encefalitis autoinmune. Se requieren estudios adicionales para establecer la terapia a largo plazo del inmunomodulador. 31,38
CONCLUSIÓN
La presentación clínica en la encefalitis autoinmune es muy variada, por lo que la aproximación diagnóstica se basa en el cuadro clínico encefalítico de inicio subagudo apoyado en hiperintensidades T2 en RM, alteraciones en EEG y hallazgos en LCR. Si bien la detección de anticuerpos antineuronales específicos confirma el diagnóstico, estos son negativos en más de la mitad de los casos. Dado que la respuesta al tratamiento también es similar en ambos grupos, comenzar la inmunoterapia en un paciente que presenta síntomas sugestivos de encefalitis autoinmune, al tiempo que descarta otros imitadores comunes, es una prioridad en al abordaje evitando retrasarse el inicio del tratamiento, lo que llevaría a un peor desenlace, deterioro progresivo y peor pronóstico.

