











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
El síndrome de Dyke-Davidoff-Masson (DDMS, según sus siglas en inglés) fue descrito por primera vez en 1933 por los investigadores Dyke, Davidoff y Masson 1.
Es considerado un síndrome epiléptico raro que se puede presentar de forma congénita o adquirida, ya sea por causas vasculares, inflamatorias, tumorales, infecciones, entre otras.
Clínicamente se puede presentar mediante crisis epilépticas, déficit motor contralateral, discapacidad intelectual, asimetría facial, trastornos del lenguaje, dificultades en el aprendizaje, trastornos sensitivos y movimientos anormales contralaterales 2.
A nivel imagenológico, sus hallazgos dependerán de la forma de presentación, ya que en los casos congénitos se observará desplazamiento de las estructuras de la línea media hacia el lado de la enfermedad y prominencia de los surcos que remplazan el tejido glótico ausente, mientras que en los casos adquiridos se observará atrofia de un hemisferio cerebral, hipertrofia ósea ipsilateral e hiperneumatización de los senos adyacentes 1.
El diagnóstico es clínico-imagenológico y dentro de los diagnósticos diferenciales a considerar se encuentran otras causas de hemiatrofia cerebral, como vasculitis unihemisférica, síndrome de Sturge-Weber, enfermedad de moyamoya, encefalitis de Rasmussen, síndrome de Silver-Russell y síndrome de Fishman 2.
Finalmente, el tratamiento es sintomático y su abordaje debe ser multidisciplinario 2.
Presentación del caso
Paciente de sexo masculino, de 46 años, procedente de Tacuarembó (departamento al noreste del Uruguay), que reside con sus padres. De su historia perinatal se destaca nacimiento a término con fórceps.
Dentro de sus antecedentes personales están: meningitis por virus herpes a los seis meses de vida, obesidad y discapacidad intelectual con dependencia total para actividades básicas e instrumentales de la vida diaria. Sin antecedentes familiares e historia de crisis epilépticas de difícil control desde los seis meses de vida hasta la actualidad, las cuales son de difícil categorización, sin un claro inicio focal y de tipo tónico clónico bilateral, según registros videográficos. El paciente presenta una alta frecuencia diaria de crisis y no ha presentado estatus epiléptico. De su historial farmacológico se destaca uso de carbama-zepina, topiramato, fenitoína, clobazam, oxcarbaze-pina y levetiracetam.
Al examen neurológico se encontró síndrome disejecutivo y síndrome piramidal de hemicuerpo izquierdo en etapa espástica, además de ausencia de elementos de síndrome neurocutáneo.
En los estudios complementarios, el paciente cuenta con electroencefalograma que evidencia actividad epileptiforme focal a derecha y episodios motores paroxísticos no epilépticos, y resonancia magnética de cráneo con protocolo de epilepsia (secuencias T2 coronal, FLAIR axial y T1 axial) que muestra una extensa disminución volumétrica en todo el hemisferio cerebral derecho, que se asocia a gliosis cortical y subcortical. Además, también se observó un engrosamiento compensatorio de la calota craneana y un aumento del tamaño del hemiseno frontal (figuras 1, 2 y 3).
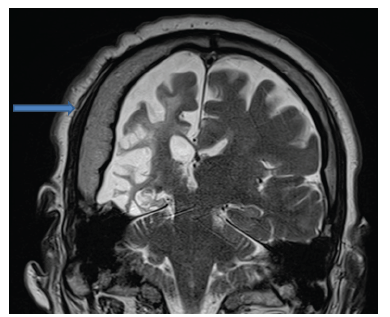
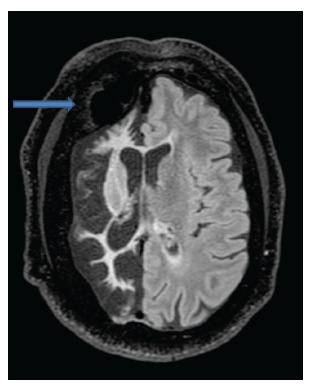
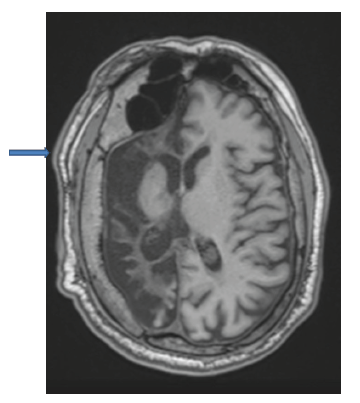
Discusión
El DDMS fue descrito por primera vez en el año 1933 en una serie de nueve pacientes que presentaban cambios radiográficos a nivel craneal en estudios de radiografía convencional y que, a nivel clínico, tenían discapacidad intelectual, crisis epilépticas, hemiparesia contralateral a la lesión y asimetría facial 1-2.
Este es considerado un síndrome epiléptico raro, siendo su prevalencia mayor en la edad pediátrica.
En las diferentes series se ha visto que es más común en el sexo masculino y que el hemisferio cerebral más afectado es el izquierdo, esto se podría explicar porque dicho hemisferio es más sensible a los cambios de la vasculatura cerebral y, a su vez, a cambios hormonales que hacen que predomine en hombres 1-3.
La fisiopatología de este síndrome sigue sin estar clara hasta la actualidad, aunque la hipótesis más utilizada es la presencia de anormalidades vasculares 2. Además, se han descrito dos formas de presentación según Alpers y Dear en 1939. Estas son: primarias (congénito) y secundarias (adquirido) 1-2.
En el caso del tipo congénito o primario, este es causado por lesiones cerebrales ocurridas en el periodo intrauterino o neonatal, debido generalmente a etiologías vasculares, mientras que en el caso del tipo adquirido o secundario es debido a infecciones, trauma, anormalidades vasculares, trastornos inmunológicos y neoplasias 1-4.
Si bien clínicamente se caracteriza por la triada de discapacidad intelectual, crisis epilépticas y déficit motor contralateral a la lesión, existen otras formas de presentación clínica como, por ejemplo, trastornos del lenguaje, síntomas sensitivos, problemas de aprendizaje, trastornos de la marcha y movimientos anormales contralaterales 2,5.
Su diagnóstico es clínico-imagenológico y es por ello que dentro de los estudios complementarios a solicitar se encuentran la tomografía de cráneo o la resonancia magnética de cráneo. Dentro de los hallazgos a encontrar están, por ejemplo, atrofia de un hemisferio cerebral, hipertrofia ósea ipsilateral, hiperneu-matización de los senos adyacentes, encefalomalacia, atrofia cerebelosa contralateral por compromiso de la vía dento-rubro-tálamo-cortical, hipoplasia de los pedúnculos cerebrales, tálamo y cápsula interna, gliosis, entre otros 2,6-7.
Dentro de los diagnósticos diferenciales a considerar se encuentran otras causas de hemiatrofia cerebral, como por ejemplo: vasculitis unihemisférica, síndrome de Sturge-Weber, encefalitis de Rasmussen, enfermedad de moyamoya, síndrome de Silver-Russell, síndrome de Fishman, entre otros 1-2,8.
El tratamiento de este síndrome es de carácter sintomático y está basado en el uso de fármacos anticrisis epilépticas. En casos puntuales se puede considerar como terapia la hemisferectomía y además se requiere de un abordaje multidisciplinario.
Finalmente, hay que señalar que el pronóstico es mejor cuando las crisis epilépticas son escasas y cuando el déficit motor aparece luego de los dos años de edad 2.
Conclusiones
Se presentó un caso del síndrome de Dyke-Davidoff-Masson de inicio en la infancia y que, a diferencia de lo establecido por la literatura, comprometió el hemisferio derecho y ha sido causa de una epilepsia refractaria.
González: escritura del borrador original, revisión y edición del manuscrito; Tammara Méndez: escritura del borrador original, revisión y edición del manuscrito.

