










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.31 no.1 Bogotá Jan./Mar. 2016
Hepatotoxicity: A Drug-Induced Cholestatic Pattern
Laura Morales M. (1), Natalia Vélez L. (1), Octavio Germán Muñoz M. MD. (2)
(1) Medical Student in the Faculty of Medicine and the Gastrohepatology Group at the Universidad de Antioquia in Medellín, Colombia.
(2) Internist and Hepatologist at the Hospital Pablo Tobon Uribe and in the Gastrohepatology Group at the Universidad de Antioquia in Medellín, Colombia.
Received: 30-01-15 Accepted: 26-01-16
Abstract
Although drug induced liver disease is a rare condition, it explains 40% to 50% of all cases of acute liver failure. In 20% to 40% of the cases, the pattern is cholestatic and is caused by inhibition of the transporters that regulate bile synthesis. This reduction in activity is directly or indirectly mediated by drugs and their metabolites and/or by genetic polymorphisms and other risk factors of the patient. Its manifestations range from biochemical alterations in the absence of symptoms to acute liver failure and chronic liver damage.
Although there is no absolute test or marker for diagnosis of this disease, scales and algorithms have been developed to assess the likelihood of cholestatic drug induced liver disease. Other types of evidence are not routinely used because of their complexity and cost. Diagnosis is primarily based on exclusion using circumstantial evidence.
Cholestatic drug induced liver disease has better overall survival rates than other patters, but there are higher risks of developing chronic liver disease. In most cases, the patients condition improves when the drug responsible for the damage is removed. Hemodialysis and transplantation should be considered only for selected cases. The effectiveness of other therapies is unproven.
This article will delve into the pathophysiology, biochemistry, and histopathology and the clinical presentation of the disease and will discuss diagnosis, management and prognosis of this type of cholestasis.
Keywords
Cholestasis, drug, liver disease, drug-induced liver disease.
INTRODUCTION
Drug Induced Liver Injury (DILI) is a rare cause of liver disease in the general population: it accounts for less than 1% of patients hospitalized with jaundice (1,2) and for up to 1% of patients managed by internal medicine (primarily with tuberculostatic and antineoplastic agents). (3) However, this entity accounts for 40% to 50% of cases of acute liver failure, (4) and it is estimated that by 6 months after onset of symptoms one in ten patients has died or required liver transplantation. One out of every five develops chronic liver disease. (5) Clearly, this entity is an important topic for research and drug monitoring. (4,6)
Three patterns of DILI have been identified: cholestatic, hepatocellular and mixed. The cholestatic pattern is characterized by levels of alkaline phosphatase (ALP) greater than twice the upper limit of normal (ULN) and/or less than or equal to 2R (R is the relationship between ALP and ALT as shown in Figure 1). The hepatocellular pattern is defined as ALT levels greater than twice the ULN and/or more than 5R while in the mixed pattern ALT is greater than twice the ULN with R from two to five. (7-10)
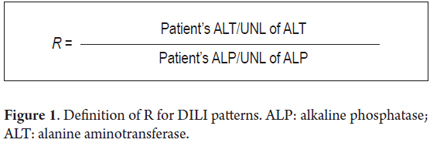
The cholestatic pattern accounts for 20% to 40% of DILI, the hepatocellular pattern accounts for 40% to 78%, and the mixed pattern accounts for 12% to 20%. (7,10) Despite the fact that there is a higher survival rate for the cholestatic presentation, the rate of improvement of patients biochemical liver profile is low and it has a high risk for development of chronic liver disease. (11)
In recent decades, multiple studies have been conducted to identify the main risk factors for development of liver toxicity as well as to determine methods of early diagnosis and proper management. This review summarizes the pathophysiological, clinical, diagnostic features of DILI and discusses the most relevant treatments.
PHYSIOLOGY
Bile is secreted through osmosis resulting from concentration of salts and other components in the bile canaliculi. Solute transport of blood to bile occurs by means of transport systems at the surface of the basolateral (sinusoidal) plasma membranes and apical canalicular membranes of hepatocytes. (12)
Basolateral membranes contains the Na+/K+-ATPase (sodium-potassium pump) and voltage-gated potassium channels (VGKCs) which have transmembrane electrical potentials of about -35 mV. This maintains the intracellular and extracellular ion gradients and pH homeostasis. This potential is what allows uptake of conjugated bile salts (bile acids) from the blood primarily by sodium-taurocholate cotransporting polypeptide (NTCP). (13) In contrast, unconjugated bile salts, organic anions and many other components which bind to albumin are transported from the plasma to hepatocytes by independent sodium transport systems such as organic anion-transporting polypeptides (OATPs). (12)
The canalicular excretion of bile acids (the major fraction of organic solutes in bile) is mediated by the family of ATP dependent transporters for bile acids and organic anions. This is the determining step for the rate of bile formation. (14) Osmotic excretion of bile acids is followed by movement of water through aquaporins and tight junctions, a flow which is dependent on bile acid. In addition to osmosis, bile acids promote canalicular secretion of phospholipids and cholesterol for the formation of mixed micelles. (13,18) There are also compounds such as reduced glutathione and bicarbonate that are independent of the flow of bile acids. Both cholangiocytes and hepatocytes secrete and absorb different components that modify the characteristics of bile as it passes through the bile duct. (Figure 2). (15)
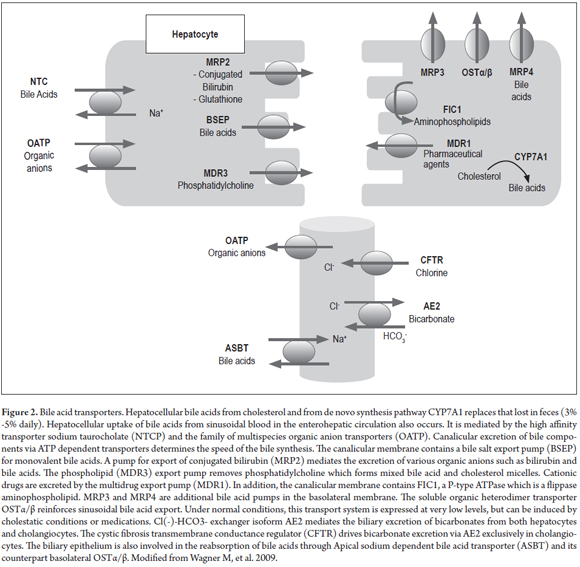
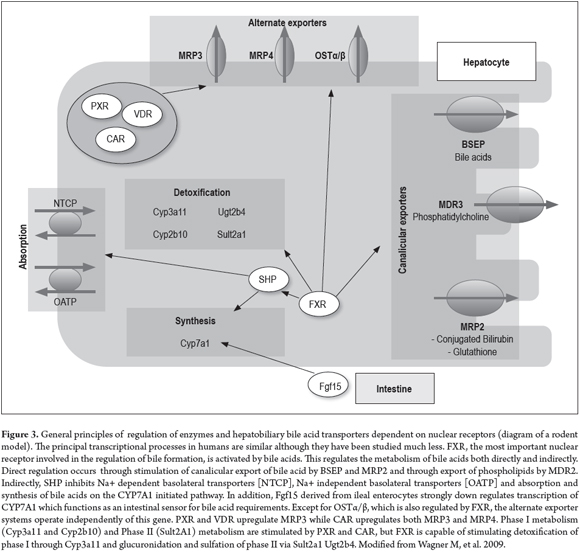
The system of hepatobiliary transporters is regulated at transcriptional and posttranscriptional levels, for example through the activation of nuclear receptor ligands. These positively and negatively regulate bile formation pathways like that of the detoxifying enzymes and pumps that export bile compounds in both pathological and physiological states. (16) Bile components, lipid products, hormones and xenobiotics work together to activate nuclear receptors such as endogenous and exogenous ligands and to modify genes that encode hepatobiliary transporters and phase I and II metabolism enzymes. (16)
There is growing evidence that the activity of nuclear receptors such as the Farnesoid X receptor (FXR) is affected by chromatin remodeling through acetylation of histones. (16) This is important because it is the best defined nuclear receptor and because it is critically involved in reducing bile acid production (CYP7A1) and in both Na + -dependent (NTCP) and Na + independent (OATP1B1 and OATP1B3) bile acid absorption. In addition, it activates monovalent canalicular excretion (BSEP - Bile Salt Export Pump) and divalent canalicular excretion (MRP2 and MDR3) of bile acid and conjugated bilirubin (MRP2) (Figure 3). (15-17, 19)
PATHOPHYSIOLOGY AND MECHANISMS OF CHOLESTASIS
Cholestasis is the basis of liver damage in every manifestation and clinical pattern of DILI. (20) In most cases, drugs cause cholestasis by inhibiting expression and functioning of hepatocellular transporters, but on rare occasion drugs induce vanishing bile duct syndrome (VBDS) which can progress to biliary cirrhosis. (21) Many cases of drug-induced cholestasis result in inhibition of liver functions resulting from the effects of the drug or its metabolites on various transport proteins and pathways. The most frequently affected, especially in cholestatic DILI, is the export of ATP-dependent bile acids via the BSEP pathway. (22) This is inhibited directly and competitively by drugs such as rifampin, cyclosporine, troglitazone and glyburide, and indirectly by metabolites of steroid hormones such as estrogen and progesterone. (23-26) The MRP2 pathway and secretion of phospholipids via the MDR3 pathway are also affected. (27,28)
Genetic alterations in the ATP-binding cassette (ABC) transporter family have also been associated with cholestatic disorders ranging from progressive familial intrahepatic cholestasis and benign recurrent intrahepatic cholestasis, to intrahepatic cholestasis of pregnancy, drug-induced cholestasis, intrahepatic cholelithiasis and biliary cirrhosis.
Homozygous alterations cause great impacts variants that result in cholestatic syndromes at early ages. (29-32) Heterozygous defects in transporters predispose to the acquisition of cholestasis through drugs, hormones and inflammation that cause decompensation of slight to moderate defects. (32)
CLINICAL, BIOCHEMICAL AND HISTOLOGICAL FEATURES
Clinical
Drug-induced cholestasis can be classified according to the anatomical site and the biochemical and histopathological pattern of the injury. These range from liver disorders that interfere with synthesis of bile to changes in bile ducts that impede excretion. (33, 34)
Usually, drug-induced cholestasis is an acute disease that quickly disappears once the agent provoking the condition is suspended. (33) It is clinically characterized by jaundice, pruritus, anorexia, malaise, nausea and fatigue. It may also have other manifestations depending on the causal mechanism and on extrahepatic drug toxicity. (35) In hypersensitive forms, systemic manifestations such as fever, rashes or eosinophilia can be seen. It is important to consider that this condition can present through a spectrum of symptoms ranging from asymptomatic biochemical abnormalities to acute liver failure. (33, 34)
Some medications cause chronic cholestasis with characteristics similar to primary biliary cirrhosis (PBC). These include xanthomas, itching and melanoderma. (35) These forms of cholestasis are considered benign because they rarely progress and can be differentiated from PBC by the absence of microsomal antibodies, acute onset of symptoms, and atypical histopathological features and clinical presentations. (33)
Biochemistry
As in other forms of cholestatic damage, primarily biochemical assessment shows elevated levels of alkaline phosphatase (ALP) and gamma glutamyl transpeptidase (GGT). (33) Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) may be normal or slightly elevated. Although bilirubin concentrations are usually high, they depend on the injury mechanism of the drug. (35)
Histopathology
Histologically, drug-induced cholestasis may occur as an acute or chronic injury which may or may not compromise the parenchyma of the liver. (35) There are two classifications of acute forms, which are more common than chronic forms.
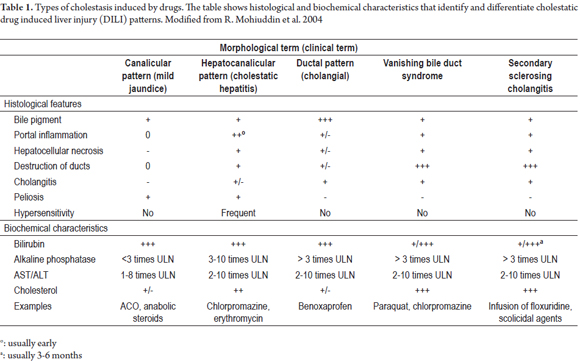
- Pure or soft cholestasis in which clots of biliary pigment are found in canaliculi which appear to be distended. Pigment accumulates in hepatocytes and Kupffer cells without inflammation or hepatocellular injury. This is more prominent in zone 3 (centrilobular area). (35, 36). Medications that cause this type include anabolic steroids, oral contraceptives and warfarin. (34, 37)
- Cholestatic hepatitis (cholangiolitis or cholestasis due to hypersensitivity) is characterized by hepatocellular compromise which is also expressed in the liver biochemistry. (35) It may be accompanied by proliferation of ducts. cholangiolitis has developed, neutrophils, lymphocytes and eosinophils will be found. (36) This pattern is common in erythromycin and chlorpromazine toxicity. (34, 35, 37)
Chronic forms of the disease last more than 6 months. Pseudoxanthomatosis develops and has a foamy appearance due to the accumulation of bile acids in hepatocytes especially in periportal regions. (33) Copper content can also be demonstrated increased with special stains and there is occasionally evidence of Mallory bodies. (37) There are also two classifications of chronic cholestasis.
- Vanishing bile duct syndrome (VBDS) initially presents as hepatocellular and bile duct inflammation but leads to ductopenia and in some cases to cirrhosis when the drug causing the condition is not suspended. It is one of the most severe presentations and can be triggered by carbamazepine, chlorpromazine, ibuprofen, amoxicillin, and clindamycin. Differential diagnosis must be done to rule out PBC and obstructive disease (35,49).
- Biliary sclerosis can occur when there is an ischemic injury of intrahepatic or extrahepatic ducts. This can simulate radiological or histological primary sclerosing cholangitis. (35,36) Medications associated with this presentation include 5-fluorodexiuridine (for the treatment of liver metastases of colorectal cancer) and formaldehyde. (35,36)
Portal area edema, infarcts and biliary lakes are late expressions of mechanical obstruction of the ducts which rarely develop in cases of DILI. (33) Nevertheless, since early histopathological presentation of biliary obstruction is indistinguishable from drug-induced cholestasis, differential diagnosis is very important. (35-38)
Table 1 summarizes the most common biochemical and histopathological features of the cholestatic patterns described. Table 2 shows drugs associated with cholestatic patterns of liver toxicity.

RISK FACTORS
Among risk factors for DILI is age which may be explained by changes in the expression of receptors and transporters, in percentage of body fat, in volume distribution or in hormonal status. (7,36) Although there are no significant differences in the incidence of DLI between genders, it has been related to changes in presentation and prognosis. The cholestatic pattern is most common among men, but the development of the disease is less favorable among women. (39) Polymorphisms and genetic factors for drug metabolism have also been identified. These include human leukocyte antigen (HLA) B * 5701, -DRB and -DRBQ haplotypes, and the MDR3/BSEP polymorphism which is associated with predisposition for pregnancy and steroid induced cholestasis. (36, 40, 50)
Alcohol use, liver disease (including steatohepatitis) and HIV infection (by a yet unknown mechanism) have also been described as risk factors. (7)
DILI has also been associated with factors dependent on increased drug strength including composition, dosage and metabolism. Examples include steroids with substitutions in C-17 (particularly alkylation or methylation), (36) doses over 50 mg/day and concomitant use of several drugs affecting the hepatic metabolism. (36, 39, 41)
DIAGNOSIS
There are no tests or markers that are absolute indicators of DILI, so diagnosis depends primarily on exclusion based on circumstantial evidence. The approach when DILI is suspected should begin with a detailed medical history including thorough questioning about medical factors, risk factors, use of prescription drugs, self-medication, and use of unconventional substances such as alternative and herbal medicine. The physician should also inquire about alcohol and other psychoactive substances. Many times this questioning should include the patients family. All these data should include the time of first use for temporal association with liver damage especially in patients with polypharmacy.
This should be complemented with the usual liver biochemical tests, standard coagulation tests, serum markers and routine imaging of bile ducts to rule out other more common causes of cholestasis (Table 3). (7, 36, 42) Other tests for autoimmune and infectious diseases may be useful for differential diagnosis. While not always necessary, a liver biopsy may be essential in selected cases, especially to assess the patients prognosis. (36)

Situations most suggestive of DILI include initiation of treatment with a new drug in the previous three months, a rash or eosinophilia, mixed type liver disease (hepatocellular and cholestatic commitment), cholestasis without alterations in imaging, acute chronic hepatitis without autoantibodies and without hypergammaglobulinemia, and patients with risk factors. The absence of any or all of these factors does not rule out the possibility of drug-induced cholestasis or any other form of DILI, but taking them into account can facilitate earlier diagnosis. (7)
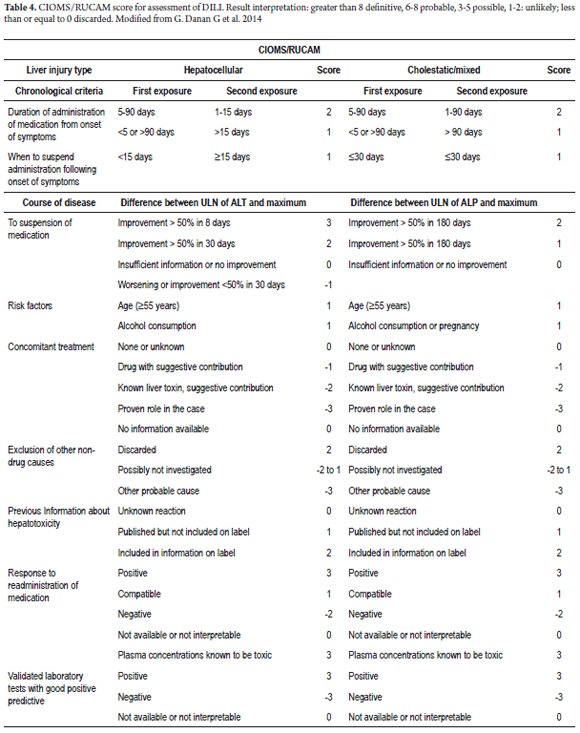
Of the several scales developed to assess the likelihood of DILI, the most widely used and best validated is the CIOMS/RUCAM scale proposed by Danan and Benichou at the International Consensus Meeting of 1990. (8) It allows classification of DILI diagnoses into definite, probable, possible, unlikely and discarded according to the pattern of liver damage (hepatocellular or cholestatic/mixed) chronological criteria, course of disease, risk factors, available information on hepatotoxicity of a drug , exclusion of other causes and response to the re-administration of the drug. This last criterion should not be used because of risks to the patient. Table 4 shows the DILI probability scale for the cholestatic pattern. Additional tests that are useful for diagnosis include the drug-lymphocyte stimulation test (DLST) and the leukocyte migration test (LMT). The first has a sensitivity of 50% and cannot unambiguously link a drug to liver damage. (43) The LMT has proven to be most useful for identifying offending substances. (44) These and other tests of this type are useful for identifying the specific drug that has caused damage, but because they are complex and expensive they are not recommended for routine use. (7)
TREATMENT
In most cases, the patient will improve when the drug responsible for the damage is removed. However, it has been found that some patients have improved even without suspension. For this reason, it is necessary to correlate the severity of the clinical picture with the importance of the use of the drug. (7)
Although there are no definitive criteria for suspension of a drug, the following have been proposed for cases of suspected cholestatic DILI because of their associations with liver damage. (7, 45)
- Bilirubin more than three times ULN.
- INR over 1.5.
Considerations for drug withdrawal in all patterns of DILI are (46):
- ALT or AST> 8 times ULN
- ALT or AST> 5 times the upper limit of normal for more than 2 weeks.
- ALT or AST> 3 times ULN with total bilirubin> 2 times the upper limit of normal or with INR> 1.5.
- ALT or AST> 3 times ULN with fatigue, nausea, vomiting, abdominal pain in the right upper quadrant, fever, rash and/or eosinophilia> 5%.
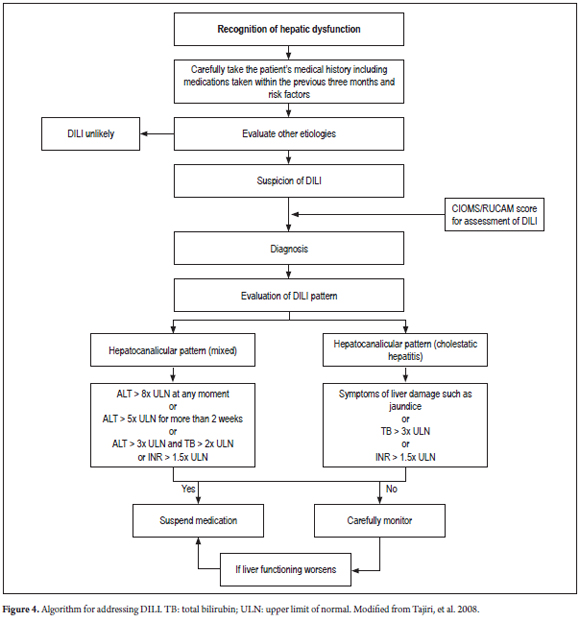
Using other medicines for managing cholestatic DILI is not supported in the literature. Although treatment with UDCA (ursodeoxycholic acid) and corticosteroids (in patients with suspected hypersensitivity) has been used, controlled clinical trials have not proven the efficacy of these treatments. (7, 46, 47, 51) Hemodialysis is rarely indicated, but whenever there is any indication of acute liver failure the patient should be hospitalized and considered for liver transplantation. (46, 47) An algorithm proposed for diagnosis and management of cholestatic DILI is shown in Figure 4.
PROGNOSIS
Information on the outcome of the cholestatic and mixed presentations is limited. It was Previously believed that the cholestatic pattern of DILI was associated with a better prognoses, but recent literature shows that 5% to 13% of these patients develop chronic liver disease (more common than in patients with hepatocellular pattern), and that 5% to 14% die or require transplantation. (36, 48)
The long-term prognosis for DILI generally depends on the clinical presentation and initial biochemistry of the patient. Aminotransferase and total bilirubin are the best predictors of mortality. Those who present ALT greater than or equal to three times ULN and jaundice (total bilirubin two or more times ULN) have a mortality rate of between 10% and 50%. This is known as the Hys Law. (48,52)
Acknowledgements
We thank Gastrohepatology group at the University of Antioquia.
Financial support
No financial support was received for this research.
REFERENCES
1. Vuppalanchi R, Liangpunsakul S, Chalasani N. Etiology of new-onset jaundice: How often is it caused by idiosyncratic drug-induced liver injury in the United States? Am J Gastroenterol. 2007;102(3):558–62. [ Links ]
2. Sgro C, Clinard F, Ouazir K, Chanay H, Allard C, Guilleminet C, et al. Incidence of drug-induced hepatic injuries: A French population-based study. Hepatology. 2002;36(2):451–5. [ Links ]
3. Meier Y, Cavallaro M, Roos M, Pauli-Magnus C, Folkers G, Meier P, et al. Incidence of drug-induced liver injury in medical inpatients. Eur J Clin Pharmacol. 2005;61(2):135–43. [ Links ]
4. Ostapowicz G, Fontana RJ, Schiødt F V, Larson A, Davern TJ, Han SHB, et al. Results of a Prospective Study of Acute Liver Failure at 17 Tertiary Care Centers in the United States. Ann Intern Med. 2002;137(12):947–54. [ Links ]
5. Fontana RJ, Hayashi PH, Gu J, Reddy KR, Barnhart H, Watkins PB, et al. Idiosyncratic drug-induced liver injury is associated with substantial morbidity and mortality within 6 months from onset. Gastroenterology. 2014;147(1):96–108. [ Links ]
6. Fontana RJ, Seeff LB, Andrade RJ, Björnsson E, Day CP, Serrano J, et al. Standardization of nomenclature and causality assessment in drug-induced liver injury: Summary of a clinical research workshop. Hepatology. 2010;52(2):730–42. [ Links ]
7. Tajiri K. Practical guidelines for diagnosis and early management of drug-induced liver injury. World J Gastroenterol. 2008;14(44):6774-6785. [ Links ]
8. Danan G, Benichou C. Causality assessment of adverse reactions to drugs-I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J Clin Epidemiol. 2014;46(11):1323–30. [ Links ]
9. Bénichou C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J Hepatol. 1990;11(2):272–6. [ Links ]
10. Bhamidimarri KR, Schiff E. Drug-induced cholestasis. Clin Liver Dis. 2013;17(4):519–31, vii. [ Links ]
11. Andrade RJ, Lucena MI, Kaplowitz N, García-Muņoz B, Borraz Y, Pachkoria K, et al. Outcome of acute idiosyncratic drug-induced liver injury: Long-term follow-up in a hepatotoxicity registry. Hepatology. 2006;44(6):1581–8. [ Links ]
12. Trauner M, Meier P, Boyer J. Molecular Pathogenesis of Cholestasis. N Engl J Med. 1998;339(17)1217–27. [ Links ]
13. Trauner M, Boyer JL. Bile Salt Transporters: Molecular characterization, function , and regulation. Physiol Rev. 2003;83(2):633–71. [ Links ]
14. Hofmann AF. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arch Intern Med. 1999;159(22):2647-2658. [ Links ]
15. Wagner M, Zollner G, Trauner M. New molecular insights into the mechanisms of cholestasis. J Hepatol. 2009;51(3):565–80. [ Links ]
16. Suchy FJ, Ananthanarayanan M. Bile salt excretory pump: Biology and pathobiology. J Pediatr Gastroenterol Nutr. 2006;43 Suppl 1:S10–6. [ Links ]
17. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A Regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517-26. [ Links ]
18. Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121(1):140–7. [ Links ]
19. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol Cell. 2014;6(3):507–15. [ Links ]
20. Ogimura E, Sekine S, Horie T. Bile salt export pump inhibitors are associated with bile acid-dependent drug-induced toxicity in sandwich-cultured hepatocytes. Biochem Biophys Res Commun. 2011;416(3-4):313–7. [ Links ]
21. Pauli-Magnus C, Meier PJ. Hepatobiliary transporters and drug-induced cholestasis. Hepatology. 2006;44(4):778–87. [ Links ]
22. Lang C, Meier Y, Stieger B, Beuers U, Lang T, Kerb R, et al. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet Genomics. 2007;17(1):47–60. [ Links ]
23. Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118(2):422–30. [ Links ]
24. Fattinger K. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: A potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69(4):223–31. [ Links ]
25. Funk C, Ponelle C, Scheuermann G, Pantze M. Cholestatic Potential of Troglitazone as a Possible Factor Contributing to Troglitazone-Induced Hepatotoxicity : In vivo and in vitro Interaction at the Canalicular Bile Salt Export Pump (Bsep) in the Rat. Mol Pharmacol. 2001;59(3):627–35. [ Links ]
26. Dawson S, Stahl S, Paul N, Barber J, Kenna JG. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos. 2012;40(1):130–8. [ Links ]
27. Smith AJ, van Helvoort A, van Meer G, Szabo K, Welker E, Szakacs G, et al. MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J Biol Chem. 2000;275(31):23530–9. [ Links ]
28. Bode KA, Donner MG, Leier I, Keppler D. Inhibition of transport across the hepatocyte canalicular membrane by the antibiotic fusidate. Biochem Pharmacol. 2002;64(1):151–8. [ Links ]
29. Sirvent A, Verhoeven AJM, Jansen H, Kosykh V, Darteil RJ, Hum DW, et al. Farnesoid X receptor represses hepatic lipase gene expression. J Lipid Res. 2004;45(11):2110–5. [ Links ]
30. Oude Elferink RPJ, Paulusma CC, Groen AK. Hepatocanalicular transport defects: pathophysiologic mechanisms of rare diseases. Gastroenterology. 2006;130(3):908–25. [ Links ]
31. Jacquemin E. Progressive familial intrahepatic cholestasis. Genetic basis and treatment. Clin Liver Dis. 2000;4(4):753–63. [ Links ]
32. Pauli-Magnus C, Stieger B, Meier Y, Kullak-Ublick G a, Meier PJ. Enterohepatic transport of bile salts and genetics of cholestasis. J Hepatol. 2005;43(2):342–57. [ Links ]
33. Mohi-ud-din R, Lewis JH. Drug- and chemical-induced cholestasis. Clin Liver Dis. 2004;8(1):95–132, vii. [ Links ]
34. Zimmerman HJ. Intrahepatic cholestasis. Arch Intern Med. 1979;139(9):1038–45. [ Links ]
35. Ramachandran R, Kakar S. Histological patterns in drug-induced liver disease. J Clin Pathol. 2009;62(6):481–92. [ Links ]
36. Bhamidimarri KR, Schiff E. Drug-induced cholestasis. Clin Liver Dis. 2013;17(4):519–31, vii. [ Links ]
37. Green RM, Crawford JM. Hepatocellular cholestasis: Pathobiology and histological outcome. Semin Liver Dis. 1995;15(4):372–89. [ Links ]
38. Watkins PB, Seeff LB. Drug-Induced Liver Injury: Summary of a Single Topic Clinical Research Conference. Hepatology. 2006;43(3):618-31. [ Links ]
39. Lucena MI, Andrade RJ, Kaplowitz N, García-Cortes M, Fernández MC, Romero-Gomez M, et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: The influence of age and sex. Hepatology. 2009;49(6):2001–9. [ Links ]
40. Andrade, RJ, Lucena MI AA. HLA class II genotype influences the type of liver injury in drug-induced idiosyncratic liver disease. Hepatology. 2004;6(39):103–12. [ Links ]
41. Lammert C, Bjornsson E, Niklasson A, Chalasani N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology. 2010;51(2):615–20. [ Links ]
42. García-Cortés M, Andrade RJ, Lucena MI, González-Grande R, Camargo R, Fernández-Bonilla E, et al. Hepatotoxicidad secundaria a fármacos de uso común. Gastroenterol Hepatol. 2005;28(8):461–72. [ Links ]
43. Takikawa H. Assessment of 287 Japanese cases of drug induced liver injury by the diagnostic scale of the International Consensus Meeting. Hepatol Res. 2003;27(3):192–5. [ Links ]
44. Usui K, Oda Y, Kubota R, Negishi K, Uno K, Tsunematsu S, et al. Clinical application of the leukocyte migration test and new diagnostic criteria for identifying causative agents in patients with drug-induced liver injury. Hepatogastroenterology. 2007;54(78):1752–7. [ Links ]
45. Food and Drug Administration F. Guidance for Industry Drug-Induced Liver Injury: Premarketing Clinical Evaluation. Center for Drug Evaluation and Research. 2009. [ Links ]
46. Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 1995;333(17):1118–27. [ Links ]
47. Norris W, Paredes AH, Lewis JH. Drug-induced liver injury in 2007. Curr Opin Gastroenterol. 2008;24(3):289-97. [ Links ]
48. Bjornsson ES, Jonasson JG. Drug-induced cholestasis. Clin Liver Dis. 2013;17(2):191–209. [ Links ]
49. Suzuki A, Brunt EM, Kleiner DE, Miquel R, Smyrk TC, Andrade RJ, et al. The use of liver biopsy evaluation in discrimination of idiopathic autoinmune hepatitis vs. drug-induced liver injury. Hepatology. 2011;54(3):931-939. [ Links ]
50. Degott D, Feldmann G, Larrey. Drug-induced prolonged cholestasis in adults: A histological semiquantitative study demonstrating progressive ductopenia. Hepatology. 1992;15(2): 244-51. [ Links ]
51. Daly P, Donaldson PT, Bhatnagar. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819. [ Links ]
52. O'Grady R, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterol. 1989;97(2):439-495. [ Links ]
