











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Idiopathic ductopenia (IAD) is a chronic condition classified within the spectrum of intrahepatic cholestatic diseases, characterized by the loss of septal and interlobular bile ductules in adulthood1. This disorder has the potential to progress to biliary cirrhosis and, in severe cases, may present as acute liver failure2. Since its first description in 1988 by Ludwig et al., the etiology remains unknown, though an inflammatory process leading to the destruction and eventual disappearance of bile ductules has been proposed. In recent years, few cases of idiopathic adult ductopenia have been reported in global literature, with even fewer in Latin American medical literature3. According to reviewed literature, two clinical presentations have been described: a mild, asymptomatic form and a more severe form in which the only effective treatment is liver transplantation. This report presents the case of a young patient from Cali, Colombia, who presented with jaundice and was ultimately diagnosed with ductopenia based on histopathological findings during diagnostic and therapeutic evaluation.
Case Report
A previously healthy 23-year-old man, with no personal or family history of liver disease, no substance abuse, or use of herbal medications, sought care at Clínica Nueva Comfenalco in Cali due to a one-week history of progressive jaundice and intractable pruritus. Blood tests revealed the following: total bilirubin (TB): 16.6 mg/dL (0-1 mg/dL), alkaline phosphatase (ALP): 349 U/L (40-129 U/L), γ-glutamyltransferase (GGT): 73 U/L (8-61 U/L), aspartate aminotransferase (AST): 67 U/L (0-38 U/L), alanine aminotransferase (ALT): 120 U/L (0-41 U/L). The patient was screened for viral infections, including human immunodeficiency virus (HIV) and hepatitis A, B, and C, all of which returned negative.
Initial suspicion was primary sclerosing cholangitis, prompting further autoimmune testing. Serological identification samples were taken for autoimmune markers, including anti-smooth muscle antibodies, anti-mitochondrial antibodies, antinuclear antibodies, asthma antibodies, antinuclear antibodies (ANA), anti-mitochondrial antibodies (AMA), anti-microsomal antibodies, liver and kidney (LKM1), all of which yielded negative results. Immunoglobulin levels were measured as follows: immunoglobulin A (IgA): 193 mg/dL (63-484 mg/dL), immunoglobulin M (IgM): 85 mg/dL (22-240 mg/dL), immunoglobulin G (IgG): 978 mg/dL (540-1822 mg/dL). Further subclass quantification revealed: IgG1: 1.31 g/L, IgG2: 0.47 g/L, IgG3: 0.1 g/L e IgG4: 0.24 g/L. Protein electrophoresis showed an alpha-1 and alpha-2 peak with a decrease in gamma A.
Leptospira infection was ruled out (negative IgM), and blood smear testing excluded hemoparasite-related diseases. Endoscopic studies showed no significant findings except for antral hyperemic gastritis. An initial abdominal ultrasound detected hepatomegaly without other abnormalities. Subsequent magnetic resonance cholangiography ruled out choledocholithiasis or biliary duct dilation. Finally, a percutaneous liver biopsy revealed marked cholestasis and loss of bile ducts in portal triads. Immunohistochemical staining confirmed focal duct destruction in portal triads (Figures 1, 2, and 3).
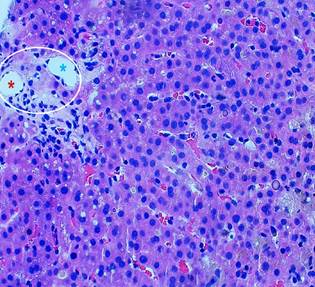
Image property of the authors.
Figure 1 The histological section shows hepatic parenchyma with a portal space (circle), where portal vessels (artery and vein) are identified but no bile duct is observed (*red: artery, *blue: vein).
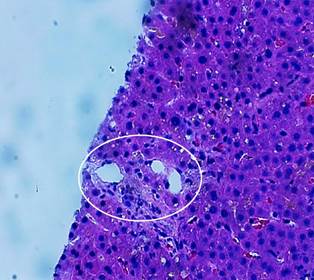
Image property of the authors.
Figure 2 The section shows hepatic parenchyma with a portal space, where a normal portal triad was identified containing portal vessels (artery, vein) and bile duct.
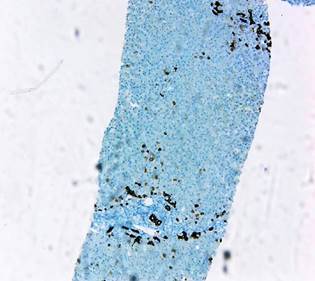
Image property of the authors.
Figure 3 Immunohistochemical study with CK7 confirming the presence of ducts and adjacent hepatocytes with biliary metaplasia, demonstrating a cholangitic injury pattern.
The patient was diagnosed with idiopathic adulthood ductopenia. Initial management with corticosteroids was initiated, achieving slow progressive reduction of bilirubin levels. For symptomatic management, cholestyramine 4 g was administered orally every 12 hours without achieving symptom control. Subsequently, ursodeoxycholic acid 300 mg was added orally every 12 hours, resulting in notable improvement.
The patient was discharged with minimal residual symptoms, with plans for continued hepatology evaluation. However, due to clinical improvement, the patient voluntarily declined further testing.
Discussion
Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) represent the most common causes of chronic hepatic cholestasis in adults4. However, a small patient subset presents with chronic cholestasis lacking antimitochondrial antibodies (suggestive of PBC) or cholangiographic abnormalities (suggestive of PSC). These cases are frequently classified as AMA-negative PBC, small-duct PSC, or idiopathic adulthood ductopenia (IAD)5.
IAD describes chronic hepatic cholestasis of unclear etiology, typically beginning in adulthood and associated with intrahepatic bile duct loss. Multiple pathogenic mechanisms for intrahepatic biliary destruction have been implicated, including biliary atresia development, immunological causes from aberrant MHC class II antigen expression, infections, ischemia, or chemical injury1-3,6. Several medications (chlorpromazine, prochlorperazine, organic arsenicals, tolbutamide) have also been associated with bile duct destruction. These were ruled out in our patient, as their identification would exclude IAD diagnosis.
Current diagnostic criteria require: biochemical cholestasis, normal endoscopic studies excluding inflammatory bowel disease, absent antimitochondrial antibodies, no granulomatous cholangitis, absent histiocytosis X, no suppurative neutrophilic cholangitis, no lymphoma or neoplasia on liver biopsy, and histopathological confirmation of ductopenia. The latter requires loss of ≥50% of septal/interlobular bile ducts in a sample containing ≥20 portal tracts, without granulomatous cholangitis or florid ductal injury1,2,6-9 (Figures 1 and 3).
Clinical manifestations vary. Some patients remain asymptomatic, though the primary symptom is jaundice with typically intense pruritus. Biochemical analysis reveals a predominantly cholestatic pattern with direct hyperbilirubinemia and elevated alkaline phosphatase/γ-glutamyltransferase1,10.
Literature describes two IAD presentations: Type 1 (often asymptomatic, may show chronic cholestasis symptoms, with less duct destruction on biopsy) and Type 2 (more severe, potentially progressing to biliary cirrhosis with extensive duct destruction histologically, where liver transplantation remains the only proven effective treatment)2,8,10,11.
For mild cases, ursodeoxycholic acid has demonstrated symptomatic benefit in multiple reports, though without documented disease progression impact. Clinical outcomes depend on ductopenia severity. Reviewed literature agrees that IAD typically progresses to end-stage liver disease when >50% duct destruction exists, while lesser involvement usually follows a benign course2,6,10. Our patient showed <50% duct abnormality on biopsy with progressive jaundice improvement. Determining whether his course will remain benign remains unclear due to potential recent diagnosis and loss to follow-up.
Conclusion
Idiopathic adulthood ductopenia is a rare cholestatic liver disease diagnosed by excluding differential diagnoses while demonstrating biochemical cholestasis and biopsy-proven bile duct loss. While recognizing this entity is important, first excluding more common pathologies remains vital for survival. When biochemical-histopathological diagnosis is confirmed, prompt management of both cause and symptoms (when possible) impacts patient survival and quality of life.

