











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Para diciembre de 2019 en Wuhan, China, surgió un coronavirus (CoV) no identificado que provocó un brote importante en muchas ciudades del país mencionado y se expandió rápidamente a nivel mundial, convirtiéndose en lo que la OMS definió como “la pandemia de la COVID-19”1. En junio 22 de 2021, el número de casos confirmados en todo el mundo había superado los 178 millones, con más de 3 millones y medio de muertes registradas2. El SARS- CoV-2 se reportó por primera vez en Latinoamérica en febrero de 2020 y posteriormente en Colombia, los primeros casos de contagios confirmados se dieron a principios de marzo de 2020 y para enero de 2021 el total era de 1’771 363 de los cuales 45 784 habían fallecido3,4.
Los CoV son virus ARN monocatenarios positivos con envoltura que tienen capacidad para infectar mamíferos principalmente5. Aunque, en la mayoría de los casos, las infecciones humanas por coronavirus son leves (como las causadas por 229E, OC43, NL63 y HKU1) es importante recordar los brotes de dos betacoronavirus como lo fueron el coronavirus del síndrome respiratorio agudo severo (SARS-CoV) en 2002-2003 y el coronavirus del síndrome respiratorio de Oriente Medio (MERS-CoV) en 2012, los cuales causaron neumonías con tasas de mortalidad de 10% y 36% respectivamente6. El virión del SARS-CoV-2 se compone de un ARN monocatenario positivo y 4 proteínas estructurales denominadas S, E, M y N7. Además de las proteínas estructurales que conforman la cápside viral, su genoma codifica varias proteínas accesorias y no estructurales (NSP) que desempeñan numerosas funciones en procesos de replicación y ensamblaje del virus8.
La caracterización y fisiopatología del nuevo CoV no está del todo esclarecida, por lo que continuamente surgen múltiples investigaciones y teorías al respecto, especialmente en idiomas extranjeros, produciendo un exceso de información no integrada y dispersa sin una aplicabilidad clara, lo cual es más evidente en el idioma español. Debido a esto, el presente artículo surge con el objetivo de realizar una revisión de tema actualizada, global y completa de la patogénesis de la COVID-19 mediante una búsqueda bibliográfica en la literatura, abordando aspectos como la caracterización del virus, el mecanismo de transmisión, su ciclo de vida, la cinética viral y la respuesta inmune del huésped, junto con la dinámica fisiopatológica de la infección en los sistemas del cuerpo más relevantes.
Metodología de búsqueda
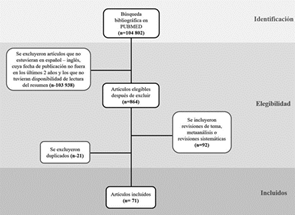
La búsqueda de la información se realizó en la base de datos PubMed entre octubre de 2020 y abril de 2021. Se usaron los términos tipo MeSH “SARS-CoV-2”, “Pathophysiology”, “Receptor binding protein”, “COVID-19”, “Autopsy findings” y “Immune response”, al igual que los términos DeCs “fisiopatología”, “respuesta inmune”, “COVID-19”, “hallazgos de autopsias”. “SARS-CoV-2” y “proteína de unión al receptor” para un total de 104 802 artículos encontrados. Se excluyeron los artículos que no estuvieran en español o inglés, cuya fecha de publicación no fuera en los últimos dos años y los que no tuvieran disponibilidad de lectura del resumen, obteniendo un total de 864 artículos. Posteriormente, se incluyeron los artículos categorizados como revisión de tema, metaanálisis o revisión sistemática para un total de 92 artículos de los cuales se excluyeron 21 duplicados. En total fueron revisados 71 artículos. (Ver Figura 1)
Desarrollo de tema
Descripción del virus
Los CoV son partículas simétricas icosaédricas envueltas, de aproximadamente 80-220 nm de diámetro y representan uno de los grupos más grandes de virus que pertenecen al orden Nidovirales, de suborden Cornidovirineae y familia Coronaviridae. Esta última se clasifica en dos subfamilias a saber, Letovirinae y Orthocoronavirinae. Letovirinae incluye al género Alphaletovirus, mientras que Orthocoronaviridae se clasifica además con base en el análisis filogenético y la estructura del genoma en cuatro géneros: Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Gammacoronavirus (γCoV) y Deltacoronavirus (δCoV)9.
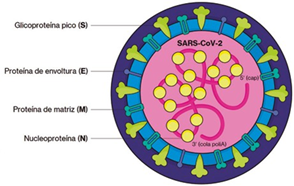
El virión del SARS-CoV-2 mide 100 nm de diámetro y está compuesto por una glicoproteína pico transmembrana o proteína spike (S) que forma homotrímeros sobresalientes en la superficie del virus, esta comprende dos subunidades funcionales (S1 y S2) y media el ingreso a las células del huésped10. También se encuentra la proteína de envoltura (E) que ayuda al ensamblaje y la liberación de los viriones, actuando como una viroporina que se ubica en las membranas celulares, formando poros involucrados en el transporte de iones; la proteína de membrana (M) es la más abundante en el virión y desempeña un papel importante en el empaquetamiento del ARN viral; las nucleoproteínas (N) corresponden a las proteínas de la nucleocápside que cumplen una función en el empaquetamiento del material genético del virus, modifican el procesamiento del ARN de la célula huésped, alteran la vía TGF-B bloqueando la apoptosis (sin embargo, también pueden inducir este proceso a través de la mitocondria), y promueven la unión del factor de transcripción NF-κB al promotor COX-2, lo que conlleva a una respuesta inflamatoria (Ver Figura 2)11,12. Además de las proteínas estructurales, el genoma codifica 16 proteínas no estructurales (NSP1-NSP16) y posiblemente, ya que el total de genes y proteínas accesorias sigue en debate, 8 proteínas accesorias mediante los marcos abiertos de lectura (ORFs) ORF3Aa ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b y ORF10 (Ver Figura 3)13.
Las proteínas accesorias poseen una baja conservación incluso entre viriones de una misma especie, complicándose la extrapolación de sus funciones por homología con otros coronavirus como se hace con el resto de las proteínas. En este sentido, se ha encontrado que la proteína ORF6 altera el transporte nuclear, y es un antagonista del IFN (interferón) tipo. Sin embargo, no es la única, se sospecha que otras proteínas como la NSP1, ORF3b, M y N también actúan como antagonistas del IFN. Mientras que, proteínas como la NSP2 y la S estimulan la respuesta mediada por IFN. ORF8 del SARS-CoV-2, la menos homóloga a las proteínas del SARS-CoV se une al MHC-I y promueve su degradación en cultivos celulares, interfiriendo con la capacidad de los linfocitos T citotóxicos en limitar la infección, que se sabe es un hecho clave para la recuperación12,14.
Mecanismos de transmisión entre humanos
La vía de transmisión comúnmente asociada al virus es por contacto directo o indirecto con los viriones presentes en gotas de origen respiratorio por parte de la mucosa nasal, conjuntival u oral. A pesar de ello, existe evidencia que apunta a la transmisión mediante aerosoles, por lo que permanecer en lugares llenos de personas y poco ventilados se convierte en un factor de riesgo. La transmisión fecal- oral y mediante fómites requieren más estudios para confirmar o descartar su papel en el contagio. No obstante, la viabilidad viral sobre superficies lisas y a bajas temperaturas y niveles de humedad, sugieren que es una posible vía de contagio15.
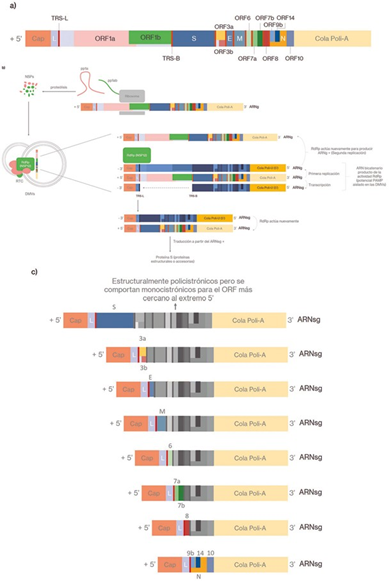
Fuente: autores. ARNsg: ARN subgenómico (no posee en su estructura al genoma completo del virus)/ ARNg: ARN genómico (posee en su estructura al genoma completo del virus)/ L: secuencia líder/ TRS-L: secuencia reguladora de la transcripción asociada a la secuencia líder/ TRS-B: secuencia reguladora de la transcripción asociada a marcos de lectura (ORFs)/ RdRP: ARN polimerasa dependiente de ARN/ DMV: vesículas de doble membrana / RTC: complejo de replicación-transcripción/.
Figura 3 Genoma, replicación y transcripción del SARS-CoV-2.
Mecanismos de patogénesis
Ciclo de vida viral e interacción con células diana
El ingreso del SARS-CoV-2 a la célula diana comienza con la unión de los viriones a la superficie celular. El virus inicialmente ingresa y se replica en las células epiteliales del tracto respiratorio superior, lo cual explica la alta carga viral localizada en esta zona y la alta transmisibilidad. Después de la adhesión, se endocita el complejo virus/receptor y la membrana viral se fusiona con la membrana endosomal permitiendo la liberación de la nucleocápside al citosol16 (Ver Figura 4).
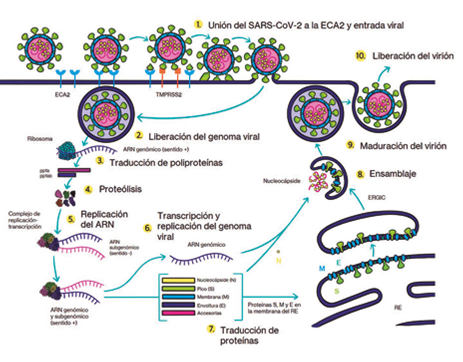
Fuente: Adaptado y traducido de Virology, transmission, and pathogenesis of SARS-CoV-2. Ilustrado por la editorial Sustancia P.
Figura 4 Ciclo de vida y replicación del SARS-CoV-2
La proteína S presente en la envoltura del SARS- CoV-2 media los procesos de unión a la célula diana y fusión de membranas requeridas para el progreso de la infección, además, altera la síntesis y modificación de lípidos13. Los análisis estructurales y bioquímicos identificaron una región de 211 aminoácidos (319- 529) en el dominio C-terminal de la subunidad S1 denominado RBD, el cual tiene un papel clave en la entrada del virus. El RBD media la unión con la enzima convertidora de angiotensina 2 (ECA2) en los aminoácidos 437-507 de la proteína S del SARS- CoV-2. Esta región en el SARS CoV2 difiere de las del SARS-CoV en cinco puntos calientes (Y455L, L486F, N493Q, D494S y T501N52). Debido a los cambios de residuos, la interacción del SARS-CoV-2 con su receptor estabiliza los dos puntos críticos de unión al virus en la superficie RBD-ECA217. Por otro lado, se ha demostrado que la proteína S del SARS-COV-2 es menos afín globalmente que la del SARS-COV a la ECA2, a diferencia de lo que sucede al comparar la afinidad de sus RBD, esto se debe a que el RBD del SARS-COV-2 en la proteína S se encuentra predominantemente en estado “lying down” (inactivo), en lugar del “standing-up” (activo) apto para la formación del complejo con ECA218.
Para ingresar a la célula, el SARS-CoV-2 requiere del procesamiento proteolítico de la proteína S, que puede ser catalizado por la serin proteasa 2 transmembrana (TMPRSS2), o la catepsina L; sin embargo, de forma distintiva puede también darse una preactivación por otra proteasa, la furina18. Esta última puede escindir a la proteína S (en la unión de las subunidades S1-S2 que permanecen asociadas ahora de forma no covalente) lo que promueve reordenamientos estructurales del RBD para la adaptación al receptor, aumentando así su afinidad19. La TMPRSS2 actúa en la membrana plasmática de la célula huésped y posiblemente durante el tráfico de vesículas endocíticas, esta proteasa se expresa en gran medida en varios tejidos y sitios del cuerpo y se coexpresa con la ECA2 en las células epiteliales nasales, los pulmones y las ramas bronquiales, lo que explica parte del tropismo tisular del SARS-CoV-2, mientras que la furina se encuentra predominantemente en pulmón, hígado e intestino delgado20. Otros receptores han sido propuestos como posibles vías alternas de entrada o factores auxiliares como el recientemente descrito GRP78 (BiP) a nivel pulmonar o el CD147 en el tejido nervioso cuya evidencia es inconclusa o insuficiente21.
Recientemente, se ha descrito otra macromolécula mediadora del ingreso viral, la neuropilina-1 (NRP1), una proteína transmembrana con actividad correceptora involucrada en la sinaptogénesis, angiogénesis y la sensación algésica. Daly et al. demostraron que la NRP1 (conocida por su capacidad de unirse a sustratos proteolizados por furina) potencia la infectividad, y por ende la propagación del SARS-CoV-222. Esta se expresa altamente en el epitelio respiratorio y olfatorio, siendo identificada en células endoteliales y epiteliales, neuronas, miocardiocitos, células musculares lisas, células dendríticas, adipocitos e incluso en la placenta. El papel de la NRP1 (en la vía dependiente de la preactivación por furina) es crítico según lo señalado en algunas investigaciones. Cantuti-Castelvetri et al. demostraron que, al bloquear una región de la NRP1, el SARS-CoV-2 reducía en gran medida su capacidad de infectar a las células. La entrada mediada por NRP1 se sugiere que es independiente de ECA2, lo que aclararía algunos casos de tropismo viral atípico que no se explican únicamente por el ingreso mediado por ECA223.
La traducción de las NSP (Ver Tabla 1) es dependiente de caperuza (cap) y se realiza mediante dos marcos abiertos de lectura en el extremo 5’; ORF1a, que origina a la poliproteína pp1a cuya posterior proteólisis da como resultado las proteínas no estructurales 1 a la 11; y ORF1b, que da como resultado a la poliproteína pp1ab de la cual derivan por proteólisis las proteínas no estructurales 1 a la 10 y 12 a la 16 (dichas proteólisis dependen de la actividad proteasa de las NSP, PLP y 3CLpro). Mientras que las proteínas estructurales y accesorias se expresan desde otros ORFs presentes en el extremo 3’ del ARN a partir de ARN subgenómicos (ARNsg)24.
Tabla 1 Función de las proteínas no estructurales (NSP1-16)
| NSP | Función |
| NSP1 | Inhibe la traducción de la célula huésped al unirse a los ribosomas de la célula* |
| Aumenta la señalización de calcineurina/NFAT (induce IL-2) | |
| Inhibe la vía de señalización por IFN-I | |
| Disminuye la fosforilación de la proteína STAT1 | |
| Modifica la función y estructura del citoesqueleto | |
| Interfiere con la replicación del ADN del huésped | |
| NSP2 | Es esencial para la estructura de los RTC anclados a las DMV |
| Interfiere con las vías de muerte y diferenciación celular | |
| Altera la síntesis y modificación de lípidos | |
| Altera el procesamiento del ADN de la célula huésped | |
| NSP3 | Mediante su actividad PLP cataliza la proteólisis de la pp1a y pp1ab |
| Posee actividad desubiquitinasa (DUB) | |
| Puede interrumpir los ciclos de la célula huésped al afectar a p53 y a la CaMKII | |
| Actividad de des-ADP-ribosilación | |
| Antagonista de IFN | |
| Participa en la formación de los RTC | |
| NSP4 | Participa en la formación de los RTC Alteración de la actividad mitocondrial |
| NSP5 | Mediante su actividad 3CLpro cataliza la proteólisis de la pp1a y pp1ab (crucial en el ciclo de vida de los coronavirus) |
| Inhibe la señalización del IFN-I al interrumpir la actividad de NF-κB y del factor de transcripción STAT1 | |
| Interfiere con la replicación del ADN del huésped | |
| NSP6 | Participa en la formación de los RTC |
| Induce formación pero restringe expansión de autofagosomas | |
| NSP7 NSP8 | Cofactores de RdRp |
| Formar un heterodímero que estabiliza el sitio de unión del ARN viral en NSP12 | |
| NSP8 tiene actividad primasa (3’-adenililtransferasa) | |
| Alteración de la actividad mitocondrial | |
| NSP9 | Participa en la replicación viral actuando como proteína de unión a ARNmc |
| Alteración del transporte nuclear | |
| NSP10 | Estimula la actividad exorribonucleasa viral (NSP14) |
| Cofactor de NSP14 y NSP16 | |
| Es un importante regulador de replicación | |
| NSP11 | Posiblemente inhibe el desarrollo de TNF-α y la señalización de IL-1 |
| NSP12 | Replica y transcribe el ARN viral (actividad RdRp) |
| NSP13 | Tiene actividad helicasa (de ADN y ARN) dependiente de magnesio |
| Actividad 5’-trifosfatasa de ARN | |
| Modifica la función y estructura del citoesqueleto | |
| Interfiere con la replicación del ADN del huésped | |
| NSP14 | Actividad exoribonucleasa (corrección de errores)** |
| Actividad N7-metiltransferasa | |
| NSP15 | Actividad EndoU sobre ARNbc y ARNmc*** |
| Retrasa la señalización del IFN | |
| Alteración del transporte nuclear | |
| NSP16 | Actividad 2’-O-metiltransferasa |
RdRP: ARN polimerasa dependiente de ARN. 3CLpro: Proteinasa similar a 3C o proteasa principal (Mpro). CaMKII: Ca2+/Calmodulina Proteína quinasa II. PLP: Proteasa parecida a la papaína o PLpro. DMV: vesículas de doble membrana. RTC: complejo de replicación-transcripción. ARNmc: ARN monocatenario. ARNbc: ARN bicatenario.
Descripción: La maquinaria de capping (aún por terminar de descifrar) se compone de el cofactor NSP10, NSP13 con su actividad trifosfatasa 5’ de ARN, NSP14 (N7-metiltransferasa), NSP16 (2’-O-metiltransferasa), faltando la identificación de la guanililtransferasa.
*Al hacerlo bloquea la entrada del ARNm celular y promueve su degradación, sin embargo, sigue permitiendo el paso y traducción del ARN viral (se cree que se media por la interacción de la NSP-1 con SL1, una horquilla del ARN viral).
**La actividad de corrección de errores (que elimina nucleótidos recién añadidos) podría explicar por qué el SARS-CoV-2 es menos susceptible a los antivirales que promueven la terminación temprana de la cadena de ARN naciente como los análogos de nucleótidos.
***Degrada la cola de poliuridinas (surgida de la replicación de la cola poli(A) del ARN viral), inhibiendo así la actividad de PAMP de estas colas, y disminuyendo la respuesta de IFN-I mediada por MDA5 fomentando la evasión inmune.
Las NSP3, 4 y 6 reorganizan la membrana del retículo endoplasmático para permitir la formación de vesículas de doble membrana (DMV), en las cuales se arma y ancla el RTC (complejo de replicación- transcripción) formado por las proteínas no estructurales NSP2 a NSP16 y la proteína N. Es allí donde a partir del ARN genómico de sentido positivo se puede producir ARN genómico de sentido negativo (ARNg-) y múltiples subgenómicos de sentido negativo (ARNsg-) por polimerización discontinua. Estos ARNsg- nuevamente se usan como plantillas para producir ARN subgenómicos de sentido positivo (ARNsg+), que una vez traducidos completan el proteoma viral. Por último, el ensamblaje de los viriones se produce en el compartimento intermedio del retículo endoplásmico-Golgi (ERGIC) para después ser liberados18.
El total de interacciones proteínas humanas- proteínas virales (interactoma) para el SARS-CoV-2 es de 332 según ha sido reportado por Gordon et al. usando solo 26 proteínas virales. Díaz J. al realizar la representación en red del interactoma del SARS- CoV-2, demuestra que el virus en mayor medida afecta los procesos de tráfico vesicular normal del huésped, y que la mayoría de las proteínas virales solo tienen una interacción o vínculo, donde solo seis proteínas tienen más de 20 interacciones, siendo las principales la M con 29 vínculos, NSP7 con 26 y orf8 con 47 vínculos. Dicha baja conectividad puede relacionarse con la alta resistencia a la deleción aleatoria de una proteína viral debido a la permanencia del resto, sugiriendo que un tratamiento combinado sobre los tres nodos principales podría ser exitoso25.
Cinética viral
Damme et al., encontraron que existe una relación dosis dependiente entre la carga viral y la gravedad de la enfermedad, por lo tanto, adquiere importancia evaluar el cambio en esta a lo largo del tiempo, lo que también es conocido como cinética viral26. Los últimos estudios demuestran que es relevante la carga total de viriones a la que se expone la persona, la cual se calcula tanto por el contacto en una sola oportunidad, como por múltiples exposiciones. Según el trabajo publicado por Harrison A. et al., el SARS-CoV-2 tenía un R0 (número básico de reproducción) de aproximadamente 2,2 (un cálculo basado en el seguimiento temprano durante el inicio de la pandemia), con un tiempo de duplicación de 5 días15. Sin embargo, la revisión sistemática de Billah et al., publicada en noviembre de 2020, reportó una media de R0 de 2.87, indicando la posibilidad de un aumento significativo de infecciones por CoV en un futuro próximo27.
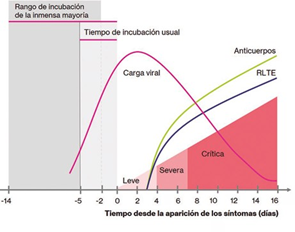
El 97,5% de los infectados que van a desarrollar síntomas, lo hacen dentro de los primeros 11,5 días a partir de la exposición (periodo de incubación). Aunque el rango de tiempo abarca desde los 2 días hasta los 14, el valor promedio es de 5-6 días, y excepcionalmente los síntomas podrían aparecer incluso después de dos semanas28.
El SARS-CoV-2 genera una amplia gama de manifestaciones clínicas que van desde una infección con síntomas leves hasta una enfermedad grave acompañada de una alta mortalidad. En pacientes levemente afectados, la respuesta inmune inicial del huésped es capaz de controlar la infección. En una enfermedad grave, la respuesta inmune excesiva conduce a daño orgánico, ingreso en cuidados intensivos o muerte (Ver Figura 5)29.
El pico viral en el tracto respiratorio se da al momento del inicio de los síntomas o dentro de la primera semana del síndrome, resaltando la capacidad del virus de colonizar y replicarse en la garganta durante la infección temprana. Algunos estudios muestran que la diseminación viral por parte del infectado como potencial vector de contagio comienza aproximadamente 2 a 3 días antes del inicio del cuadro clínico, o en los primeros 5 días desde la aparición de los síntomas. Después de la primera semana de las manifestaciones clínicas, no ha sido documentada la transmisión viral. La relación entre el síndrome y la carga viral del SARS-CoV-2 es una de las razones que favorecen su infectividad comparada a otros betacoronavirus como el SARS-CoV que posee una disociación más acentuada entre el síndrome (de forma más temprana) y el pico de la carga viral (más tardía), lo que facilita la autopercepción de enfermedad y el aislamiento consecuente29.
Respuesta inmune celular
En el momento de la exposición, el virus es detectado por receptores del sistema inmune innato (PRR) como el RIG-I (gen I inducible por ácido retinoico) y los receptores endosomales tipo toll (TLR) que fomentan la producción de mediadores proinflamatorios. Los ARN de doble cadena son reconocidos tanto por el RIG-I como por el MDA5 (proteína 5 asociada a la diferenciación de melanoma), que una vez activados se unen con la proteína adaptadora mitocondrial MAVS, conduciendo a una cascada de señalización que activa los factores de transcripción IRF3 (factor 3 regulador del interferón) y NF-κB. Esto aumenta la expresión de los IFN tipo I (dentro de los que se encuentran el IFN-α y el β) y III (IFN-λ). Los IFN posteriormente pueden unirse a su receptor señalizando de forma autocrina o paracrina mediante la vía JACK-STAT, lo que incrementa la transcripción de productos de genes estimulados por IFN (ISG), y de igual forma, aumenta la expresión de estos INF, interfiere con la replicación viral y fomenta la activación de macrófagos y células antivirales como los linfocitos NK. Se considera que dicho feedback positivo puede ser uno de los substratos para que la respuesta originalmente paracrina/autocrina derive en una respuesta inflamatoria sistémica, sin embargo, los bajos niveles de IFN encontrados a nivel periférico en casos graves parecen contrariar esta hipótesis30.
La acción antiinflamatoria reportada de los IFN se puede explicar por su capacidad de limitar la replicación viral y restringir la respuesta inflamatoria sistémica31. El SARS-CoV-2 se caracteriza por la evasión a la detección temprana por parte del sistema inmune, en el cual la señalización y la producción de IFNs se encuentra alterada. El nuevo CoV produce una potente supresión de la expresión de IFN tipo I y III a través de las proteínas virales, y evasión de la detección mediada por RIG-I debido a la compartimentalización de la reproducción viral (en las DMV). La actividad RIG-I ya se encuentra suprimida en los pacientes ancianos, lo que representa uno de los factores que contribuyen a una mayor mortalidad y severidad en este grupo etario32.
En los casos más graves se observa una respuesta inflamatoria desproporcionada del huésped con altas concentraciones de citocinas y quimiocinas proinflamatorias, y menores niveles de IFN-β o IFN-λ a los esperados, lo que da como resultado una viremia persistente, replicación exagerada y la posterior respuesta hiperinflamatoria. Estas moléculas proinflamatorias en alta concentración amplifican el daño tisular a través de la disfunción endotelial y la vasodilatación, permitiendo el reclutamiento de células inmunes, como linfocitos además de macrófagos y neutrófilos responsables del desarrollo de lesiones pulmonares, pudiendo culminar en insuficiencia respiratoria, insuficiencia orgánica y muerte33. Así mismo, la COVID-19 grave se caracteriza por alteraciones como la pérdida de células dendríticas plasmocitoides, basófilos y una linfopenia T muy marcada de predominancia CD8+ y γδ34.
Ante la infecciónseproduceuna respuesta coordinada tradicional por linfocitos T CD4+ y CD8+, cuya actividad se ha visto en algunos casos aumentada y en otros disminuida35. Cuando la respuesta linfocitaria es insuficiente se cree que se vincula con la severidad de la enfermedad, un ejemplo claro es la linfopenia que representa un factor de riesgo asociado con la admisión a unidad de cuidados intensivos, severidad y mortalidad36. Las células T CD8 + específicas del SARS-CoV-2 están presentes en aproximadamente el 70% de los pacientes que se han recuperado37. Un estudio ha demostrado que la seropositividad no está completamente vinculada a la respuesta adaptativa celular, siendo que aproximadamente 93% de los “asintomáticos” presentaron respuesta celular específica por linfocitos T ante el SARS- CoV-2 a pesar de la seropositividad en solo el 60% de los casos38. Cabe destacar que en el momento de activarse la respuesta inmune celular se incrementará la producción de mediadores inflamatorios (IFN-I, TNF-β, IL-1, CCL2, IFN-α, IL-1β, IL-6, TNF-α, CCL5), la perforina y granzima B, procesos que suelen ocurrir en otras infecciones de las vías respiratorias39.
Respuesta inmune humoral
La mayoría de los anticuerpos neutralizantes (Ac- nt) contra el SARS-CoV-2 se unen al RBD, y aparecen dentro de los primeros 20 días desde el comienzo de los síntomas en la mayor parte de los casos. La magnitud de la respuesta humoral (tanto los anticuerpos como los linfocitos B de memoria anti- RBD) dependen positivamente de la severidad de la enfermedad sin afectar la cinética humoral, de hecho, los títulos más elevados se presentan en los pacientes hospitalizados más graves comparados a los menos afectados, y además, en los pacientes mayores y de edad media comparados a los más jóvenes40. Además, un porcentaje considerable (>30%) de los infectados se recupera con niveles bajos o incluso sin desarrollar Ac-nt, y se ha encontrado una asociación negativa entre los títulos de anticuerpos y el conteo de linfocitos al momento de la admisión41.
Los títulos de Ac-nt leves tienden a desaparecer más rápidamente (incluso en periodos tan cortos como 50 días para los menores títulos) que los títulos elevados, aun cuando los anticuerpos de unión puedan seguir detectables. Comparativamente, se sugiere que la memoria mediada por linfocitos T podría ser más estable o decaer más lentamente. Por otra parte, no se tiene seguridad de que la inmunización sea esterilizante, y por lo tanto existe la posibilidad de que personas inmunes actúen como vectores de contagio42.
La duración de los anticuerpos contra el SARS-CoV-2 en plasma sigue en estudio, Gaebler et al., demostraron la persistencia de estos después de 6 meses desde la infección en individuos recuperados, sin embargo, la actividad neutralizante en plasma disminuyó sustancialmente, al mismo tiempo que los títulos de anticuerpos anti-RBD donde las IgA fueron las menos afectadas43. Además, evidenciaron que la magnitud de la respuesta por células de memoria contra el RBD viral permaneció prácticamente constante (aunque los anticuerpos que estas expresaban mejoraron su potencia neutralizante). Contrariamente, Dan et al., encontraron que aproximadamente después de 3 meses los niveles de IgA anti-RBD eran indistinguibles en los infectados vs los no infectados, los anti-RBD tipo IgG fueron potencialmente estables y que los niveles de linfocitos B de memoria incrementaron en la mayoría de los casos hasta 8 meses después de la aparición de los síntomas para una corte que se componía mayormente de casos leves y algunos hospitalizados40.
Por otro lado, el inmunofenotipo humoral descrito en una cohorte de Singapur demostraba que los títulos de anticuerpos neutralizantes eran comparables entre pacientes asintomáticos y sintomáticos. Además, la fuerza y especificidad antigénica de la respuesta celular T no mostró mayores diferencias entre pacientes asintomáticos y sintomáticos, lo cual parece indicar que la magnitud de las respuestas B y T no se correlacionan con el resultado clínico. Por otra parte, ambas clases de pacientes montaron además una fuerte respuesta citoquímica ante estimulación de células de sangre completa con péptidos virales, sin embargo, los pacientes asintomáticos tendían a expresar altas concentraciones de IL-2 e IFN-γ, contrario a las bajas concentraciones de estas citoquinas en los sintomáticos44.
“Tormenta de citoquinas”
La tormenta de citoquinas o síndrome de liberación de citoquinas es una respuesta inmunológica mal direccionada, exagerada y desregulada que se acompaña de una excesiva expresión de citoquinas y quimiocinas proinflamatorias por parte de células inmunes y no inmunes. Esta, para generarse, no requiere de un sistema inmunológico originalmente desregulado, de hecho, un sistema óptimo puede reaccionar de forma excesiva45. La liberación masiva de citoquinas proinflamatorias contribuye a la inflamación y el daño pulmonar extenso llevando a la lesión pulmonar aguda, el SDRA y la neumonía fatal; además de alteraciones inflamatorias en otros sistemas, entre las que se encuentran la falla multiorgánica, linfopenia severa sin necesidad de infección directa y pérdida de linfocitos Th reguladores con ganancia de Th17, disbiosis intestinal, incremento de la permeabilidad vascular, hipercoagulabilidad y trombosis. Estos eventos se acompañan de otros potenciadores del daño mediados por el virus, como la activación del complemento, de células proinflamatorias, y la formación de autoanticuerpos, células inmunes autorreactivas y complejos inmunes46.
En la aparición de este síndrome participan múltiples citocinas proinflamatorias producidas por numerosas células como células epiteliales, macrófagos, linfocitos T, neutrófilos y células Th17, incluidas TNF-α, IL-6, IL-8, IL-17 e IL-1β, además de quimiocinas proinflamatorias como CXCL10 (IP-10 o proteína 10 inducida por IFN-γ), y CCL2 (MCP1)46,47. Algunos patrones de expresión tienen un papel pronóstico central, como la expresión de la triada IL- 6, IL-10 e IP-10, que se correlacionan cercanamente con la progresión de la enfermedad, donde la IP-10, a diferencia de en otras infecciones virales, permanece elevada a lo largo de la COVID-19, y en este caso en particular su expresión parece ser independiente del IFN-γ. Así mismo, lo que parece ser un sello hiperinflamatorio de la COVID-19 es el incremento de respuestas de IFN tipo II y citoquinas mediadas por la vía NF-κB, a su vez que se suprimen las respuestas de IFN tipo I y III, que poseen un papel antiviral particularmente importante para el control viral en las fases iniciales. Esto se pone en evidencia por la correlación con severidad que existe en pacientes con variantes que afectan la respuesta interferónica tipo I, o autoanticuerpos contra dichas moléculas44. Por otro lado, existen estudios que demuestran que la acción sinérgica del TNF y el IFN-γ (IFN tipo II) median la PANoptosis (Piroptosis, Apoptosis y Necroptosis) asociada con la inducción y perpetuación de la tormenta de citocinas y la muerte celular48.
La unión de los viriones del SARS-CoV-2 a la ECA2, su internalización e inhibición mediada por ADAM17, conlleva a un incremento en la actividad de la angiotensina II/NF-κB induciendo la liberación de citoquinas como IL-6, IL-8 y TNF-α, que también se ven expresadas por la activación de receptores intracelulares y por procesos como la piroptosis, una muerte celular inflamatoria, vinculada a la actividad del inflamosoma y la IL-1β, actividad que las viroporinas E y ORF3a son capaces de inducir. Este proceso podría representar un vínculo entre la respuesta inflamatoria grave y la pérdida del control sobre la replicación viral. Además, los patrones de citoquinas como el de IL-1β, IL-23, IL-6 y TGB-β promueven la transcripción de RORγt, que junto a la hipoxia del entorno celular inducen la diferenciación de los linfocitos T CD4+ en TH17, productores de IL-17, favoreciendo a este grupo celular por sobre los linfocitos Th reguladores. Por otro lado, la IL-6 induce la activación del factor NF-κB y producción de otras citoquinas como TNF-α, y el incremento en la expresión de IL-8 mediado por la activación de las vías JNK, p38 o ERK inducidas por la infección viral, junto a la IL-17 reclutan y activan neutrófilos que favorecen el daño celular y tisular. Los efectos mediados por las citocinas no son aislados, debido a que estas se interregulan. Ejemplos de estos vínculos son la inducción de IL-6 e IL-8 por la IL-17, o de NO, PAF, PGE2 e IL-6 por parte de la IL-1β, o el reclutamiento de neutrófilos e inducción de IL-1β e IL-6 dado por la actividad TNF-α47.
Dinámica fisiopatológica de la infección por sistemas principales
Sistema hematológico
Las manifestaciones hematológicas graves de la COVID-19 tienen características tanto de una microangiopatía trombótica (MAT) como de coagulación intravascular diseminada (CID). Los hallazgos histopatológicos de los exámenes post mortem en pacientes con la COVID-19 mostraron ser compatibles con los de una MAT por los depósitos plaquetarios microvasculares típicos en vasos pequeños de los pulmones a lo largo de los focos de hemorragia local y la acumulación de células inflamatorias en los capilares alveolares49. Sin embargo, la afectación hematológica de la COVID-19 no presenta la totalidad de características de la MAT, como lo son la hemólisis intravascular y la trombocitopenia severa, debido a lo cual diversos autores afirman que se trata de una MAT con compromiso local a nivel pulmonar50.
Como en la CID, los pacientes con la COVID-19 generalmente muestran un aumento de los niveles de dímero D y fibrinógeno, no obstante, se han presentado niveles muchos más bajos de antitrombina, proteína C y de los factores de coagulación (II, V, VII, X), a los que normalmente se presentan en la CID. Adicionalmente, solo el 5% de los pacientes fallecidos registraron los criterios establecidos por la Sociedad Internacional de Trombosis y Hemostasia para CID, y ninguno de los sobrevivientes cumplió con todos los criterios51.
La manifestación clínica de la coagulopatía por COVID-19 es principalmente protrombótica en ausencia de una coagulopatía de consumo real, con una alta tasa de tromboembolismo venoso y posiblemente arterial con pocas complicaciones hemorrágicas, por lo que recientes estudios concluyen que debe clasificarse como una manifestación distinta de un síndrome de coagulación intravascular que necesita de nuevos criterios diagnósticos52.
Sistema respiratorio
Los pacientes que desarrollan las formas severas de la enfermedad demuestran hallazgos histopatológicos similares a los encontrados en el Síndrome de Dificultad Respiratoria Aguda (SDRA), con elevada infiltración linfocítica intersticial e invasión intraalveolar de macrófagos. Un hallazgo distintivo en la COVID-19 es la presencia de tapones producto de exudados mucofibrinosos en el tracto respiratorio, asociados a las citoquinas proinflamatorias y que se relacionan con los casos de severidad incluso en los adultos jóvenes53.
La enfermedad a nivel pulmonar puede pasar por tres etapas según los hallazgos histopatológicos; la primera caracterizada por el daño alveolar difuso (DAD), que junto a los cambios reactivos de los neumocitos puede encontrarse incluso antes de la aparición de los síntomas. La siguiente etapa es la trombosis difusa de arterias pulmonares pequeñas que se halla con frecuencia de forma temprana después del inicio de los síntomas; y finalmente, la fibrosis intersticial, que comienza su aparición aproximadamente a las 3 semanas desde el inicio del cuadro, sin relación aparente con la ventilación mecánica54. Los hallazgos se llegaron a presentar de manera simultánea, y las tres etapas no siempre se dieron5.
El fenómeno de hipoxemia silenciosa o “feliz” consiste en una hipoxemia arterial pronunciada, sin síntomas proporcionales al inicio de la enfermedad como la disnea. En la COVID-19 múltiples cambios pulmonares llevan al desarrollo de hipoxemia. Inicialmente, se encuentra vasoplejía asociada a la infección que favorece el flujo sanguíneo (vasodilatación persistente) en alveolos colapsados debido a la pérdida de surfactante en el entorno inflamatorio y al edema intersticial, así mismo, se presenta vasoconstricción que limita el flujo en otras zonas que pudiesen estar mejor ventiladas. Entre otros factores prohipoxémicos están los microtrombos intravasculares, la formación de membranas hialinas y disminución de la hematosis. Como en los primeros días de la enfermedad, la compliancia pulmonar, el espacio muerto fisiológico y la resistencia en la vía aérea se encuentran normales, no se producen estímulos prodisneicos importantes. Es por esto que, la taquipnea, la hiperpnea y el aumento esperado del gradiente de oxígeno alveolar-arterial deben tenerse en cuenta al momento de interpretar valores de SpO2 “normales”, pues estos pueden corresponder a valores producto del desplazamiento a la izquierda de la curva de la disociación de la hemoglobina56.
Sistema cardiovascular
Se ha demostrado que la ECA2 se expresa en miocitos cardíacos humanos, por lo que es posible que el SARS-CoV-2 infecte directamente a las células cardíacas, e induzca miocarditis. Se ha planteado un daño indirecto mediante 4 mecanismos principales: la injuria isquémica, la coagulación anormal, la alteración de la microcirculación y la tormenta de citoquinas57.
Actualmente, entre el 7,2% y 40,9% de los pacientes que cursan con la COVID-19 desarrollan lesión miocárdica. Los biomarcadores que frecuentemente se alteran son la troponina cardíaca y el péptido natriurético tipo B, además de la disminución de la fracción de eyección ventricular izquierda. Estos hallazgos, se pueden corresponder con las descripciones histopatológicas, donde se ha llegado a reportar infiltrado linfocítico, edema miocárdico como parte de una miocarditis leve, signos de epicarditis y endotelitis cardiaca55.
Además de las citoquinas proinflamatorias como IL-6 y TNF-α, la piroptosis asociada a la infección directa se ha encontrado como mediador de la disfunción endotelial. Este proceso, junto a la disrupción del glicocálix, y endotelitis por la leucotaxis conllevan a una actividad procoagulante. Se especula que la piroptosis se puede dar mediante tres mecanismos que ensamblan el inflamosoma: la acumulación de angiotensina II, la reacción del complemento ante la proteína N, o por unión directa de la proteína S con la ECA2. Una vez activado, se producen citoquinas proinflamatorias (IL-1β e IL-18) y se liberan DAMPs (parones moleculares asociados al daño), lo que lleva a la activación de la cascada del complemento y la subsecuente cascada de la coagulación52.
Sistema neurológico
A pesar de que la detección de ARN viral es baja en el SNC, existe una fuerte evidencia de que los coronavirus también invaden el SNC pudiendo explicar diferentes neuropatologías y daños focales como el del centro cardiorrespiratorio55. Algunos estudios también indicaron la presencia de partículas de SARS-CoV-2 ubicadas en neuronas del cerebro en muestras de individuos infectados58. Otras investigaciones sobre coronavirus sugieren que la diseminación neuronal retrógrada y hematógena pueden ser las posibles vías para que el SARS-CoV-2 ingrese al SNC59. Además, Belani P et al, afirman que la COVID-19 representa un factor de riesgo para los accidentes cerebrovasculares isquémicos60.
Se ha encontrado mayor representación del ARN viral en el bulbo olfatorio que en el tallo cerebral61. La anosmia es el síntoma neurológico principal, e incluso hay quienes consideran que es de mayor utilidad que la fiebre como criterio de tamizaje, siendo uno de los indicadores más tempranos comúnmente reportados de la COVID-19, algo que se ha vuelto cada vez más incierto con los cuadros sindromáticos predominantes ante las nuevas variantes virales. Esta puede deberse a un efecto directo del SARS-CoV-2 sobre las células sustentaculares (CS), o indirecto, al causar una disfunción metabólica secundaria u otra disfunción de las neuronas receptoras olfativas, ya que las CS también sirven para proteger estas neuronas. Las CS se regeneran después del daño con una frecuencia más rápida que las neuronas receptoras, lo que explica por qué la anosmia asociada a la COVID-19 suele ser de corta duración, sin embargo, algunos estudios reportan casos de anosmia que perdura por varios meses, e incluso parosmia, que se cree está asociada a la regeneración del epitelio olfatorio62. Aunque la prevalencia es muy variable, se puede encontrar disfunción olfatoria hasta valores cercanos al 80%. Además, se han encontrado ageusias verdaderas que se independizan de las relacionadas con la pérdida del olfato63.
Sistema renal
En el sistema renal, la necrosis tubular aguda proximal fue la lesión más frecuente encontrada en autopsias, si bien, su etiología aún no está bien definida, parece ser multifactorial, involucrando mecanismos relacionados con la alta liberación de citoquinas, la hipoxia sistémica, alteraciones de la coagulación, sepsis, o incluso la destrucción celular debido a la virosis64. En otros hallazgos post- mortem la proteína N del SARS-CoV-2 se encontró acumulada en los túbulos renales, lo que sugiere que infecta directamente a través de diseminación virémica induciendo lesión renal aguda al unirse a la ECA2 e internalizarse y por lo tanto infectar a las células renales que lo expresan, incluidas las células epiteliales tubulares renales, los podocitos y otros65.
Sistema reproductivo
La COVID-19 podría generar infertilidad masculina y deficiencia de testosterona. A nivel de las células testiculares, cuatro células principales expresan una mayor tasa de ARNm de la ECA2, siendo superior en varones jóvenes: células de ductos seminíferos, espermatogonias, células de Leydig y las células de Sertoli66,67,68. La infección directa y las consecuencias inflamatorias por citoquinas representan los mecanismos patológicos sobre las células de Leydig que podrían llevar a una disminución de la testosterona sérica69.
Hasta la fecha, no se han informado daños al sistema reproductor femenino en pacientes con la COVID-19. Sin embargo, la alteración del sistema renina angiotensina aldosterona (SRAA) asociada a la infección por SARS-CoV-2 afecta la foliculogénesis, esteroidogénesis, maduración de ovocitos y ovulación pudiendo resultar en infertilidad y trastornos menstruales70. Cabe mencionar que la expresión de ECA2 en la placenta es mayor que la detectada en pulmón por lo que existe riesgo de infección viral71.
Conclusiones
Con el presente artículo se logró realizar una revisión de los hallazgos más actualizados en la literatura científica respecto a la caracterización y fisiopatología del SARS-CoV-2 junto con la dinámica de infección en los sistemas principales, siendo fundamental este conocimiento para la identificación de blancos terapéuticos, el entendimiento del comportamiento de la enfermedad y el desarrollo de vacunas.
En cuanto a la respuesta inmune, la posible evasión temprana representa una paradoja al tomar en cuenta la hiperinflamación posterior, donde la disrupción interferónica a nivel pulmonar juega un papel importante posibilitando la extenuación de linfocitos o la generación de respuestas autoinmunes que deberían evaluarse meticulosamente por la presencia de linfopenia. Para contrarrestar la evasión temprana, se podría considerar como un posible método terapéutico iniciar tratamiento con IFN buscando evitar la replicación excesiva.
Por otra parte, se debe pensar en la COVID-19 no meramente como una enfermedad pulmonar, sino como una enfermedad multisistémica resaltando sus efectos a nivel hematológico, neurológico y renal en donde el mecanismo y el alcance de lesión sobre ellos aún no están del todo descritos. Vale la pena resaltar las alteraciones hematológicas de la enfermedad las cuales abren la posibilidad de establecer nuevos criterios para lo que parece ser una CID modificada asociada a la COVID-19. Así mismo, respecto a la asociación clínico-patológica en el sistema respiratorio, es importante poder identificar la presencia de hipoxemia feliz, dado que plantea el escenario para un deterioro clínico rápido asociándose a una mayor internación en UCI y mortalidad intrahospitalaria.

