











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Familial Hypercholesterolemia (FH) is an autosomal genetic disease homozygous (HoFH) or heterozygous (HeFH) with an important impact on quality of life and life expectancy. It is characterized by high low-density lipoprotein (LDL-C) levels and early-onset atherosclerotic cardiovascular disease (ACVD), which is more premature in HoFH given the high LDL-C burden since childhood1. In order to suspect the disease, it is necessary to ask about family history of hypercholesterolemia and major cardiovascular events before the age of 50. Awareness of this disease is increasing, as prompt management could significantly modify adverse health outcomes. FH globally affects 14 to 34 million people, this data is an underestimation because only 22/200 nations have reported FH general prevalence, and just ~1 % of the total cases in each country has been registered. FH does not have an ICD-10 code independent of other types of hyperlipidemia, making it difficult to construct reliable registries2 3.
Vallejo-Vaz et al, in 2018 reported a global HeFH prevalence of 1:250-500 individuals; while the prevalence of HoFH in Europe is much lower, 1:160 000-860 0003,4. The Ibero-American FH Network (IBAFH) has estimated that, in Ibero-America, 1.2-2 million cases of HeFH and 600-1800 of HoFH exist, but only 27 400 cases have been diagnosed. Genetically classifying those affected is important because HoFH leads to early-onset ACVD, which could be prevented with lipid-lowering treatment5. These data may be higher due to the limited availability of molecular tests to classify FH cases5.
In Colombia, there are no FH populational studies; but case reports and case series in which the molecular variants involved in FH are identified. Rincón et al presented in 2018 two FH cases with poor response to lipid-lowering treatment, who carried a homozygous deletion in the LDLR gene6. Ruiz et al. published 36 FH cases, 34 of which undergo molecular testing: 33 had at least one mutation in one allele in the LDLR gene, 14 were homozygous, 12 were compound heterozygous, two were double heterozygous, and five subjects were single heterozygous7. López et al. in 2018 carried out an LDLR gene mutational analysis in a cohort of 27 individuals belonging to 24 families with FH; among all mutations, three pathogenic variants were detected8. In the mentioned Colombian cases, only in one of them, the variant c.-135C>G in the LDLR gene was identified, this variant was the one described in the cases we report here. This variant is pathogenic according to ClinVar, Ensembl, and LDLR-LOVD databases. In ClinVar it was reported ten times: six times it was classified as probably pathogenic and four as pathogenic. The LDLR-LOVD database has five reports of this variant, in four appears as probably pathogenic and in one as pathogenic. In Ensembl, the variant is identified as rs879254375 and has two reported alleles c.-135C>G and c.-135C>A. All databases define it as a germline mutation associated with hypercholesterolemia that has an autosomal dominant inheritance pattern.
The decrease in the LDLR number, secondary to this mutation, explains the high LDL-C. This variant is located in the promoter 5' to the open reading frame and prevents the proper coupling of the transcription enzymatic machinery to DNA, leading to a decrease of the LDLR transcription9 (Figure 1). When this mutation is in heterozygosity, the other allele produces the LDLR mRNAs. The result is a significant reduction in LDLRs and the accumulation of LDL-C in blood. When homozygous for this variant, the number of LDLRs are almost null, affected individuals usually have LDL-C> 500 mg/dL, and exhibit a lower response to treatment.
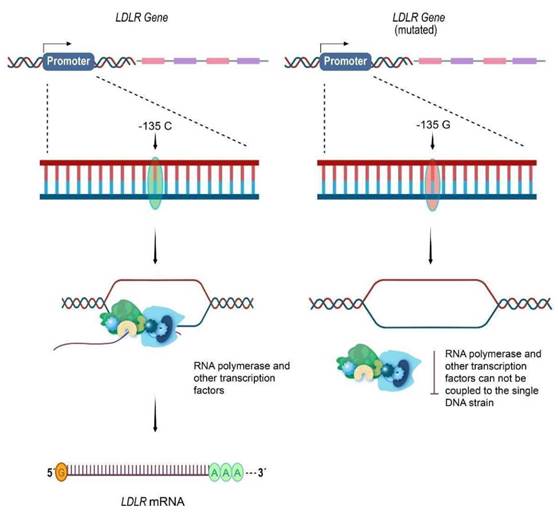
Source: authors.
Figure 1 Molecular pathophysiology related to LDLR promoter mutation c.-135C>G. Right: When the LDLR gene promoter has the pathogenic variant c.-135C>G, it prevents the RNA polymerase II and other transcription factors from properly coupling to the open DNA strand. This leads to the absence of the mRNA and a decrease in the LDLR number. Left: The non-mutated allele of the LDLR gene goes through the normal transcription process. The LDLR mRNA is generated and subsequently traduced in the ribosomes to LDLR.
FH treatment includes healthy lifestyles and pharmacological management. Weight loss, smoking cessation, and aerobic exercise are important, but they rarely achieve LDL-C goals; making lipidlowering drugs necessary4. This disease requires a holistic approach in order to achieve adequate levels of LDL-C, taking into account physical activity, diet, and pharmacological therapy. Molecules can be used for monotherapy or in combination, for instance, statins promote the expression of LDLR on the cell surface, thus, moving LDL-C inside the cells.
Herein, we report a family with HeFH caused by the pathogenic molecular variant c.-135C>G in the LDLR promoter, highlighting the significance of FH cascade diagnosis and molecular classification of disease's zygosity to establish early comprehensive management.
Case Description
Case one/III9
54-year-old female with hypercholesterolemia, hypertension, and dilated heart failure; physical exam revealed bilateral corneal arcus, no xanthomes, nor xanthelamas. At age 10 her LDL-C was above 300 mg/ dL, but she didn't receive treatment or systematic follow-up. At 21 she was found again to have elevated LDL-C levels (>300 mg/dL), so she began lifestyle modifications and lipid-lowering treatment with lovastatin, which was suspended due to myalgia. In the following 20 years, her LDL-C remained >300 mg/ dL on occasional follow-ups. Still, no medications were provided.
At age 40, she presented an Acute Myocardial Infarction (AMI) and underwent Percutaneous Transluminal Coronary Angioplasty (PTCA) for implantation of a stent in the Left Anterior Descending Artery (LADA). Her LDL-C was 460 mg/ dL, so after discharge, she resumed treatment with lovastatin, promptly re-suspended without achieving LDL-C goals.
At age 44 (LDL-C 380 mg/dL), she suffered another AMI that led to placement of a second stent in the LADA. She also developed Heart Failure (HF) with reduced Left Ventricular Ejection Fraction (LVEF). After discharge, she initiated treatment with antihypertensives, statins, anticoagulation, and beta-blockers (see table 1). Hypertension control was achieved, but LDL-C remained over 300 mg/dL.
At age 48, she had a stroke in the left middle cerebral artery territory. In the brain images, besides the acute involvement, other old infarction areas were observed. She had a motor sequel that partially improved with physical therapy.
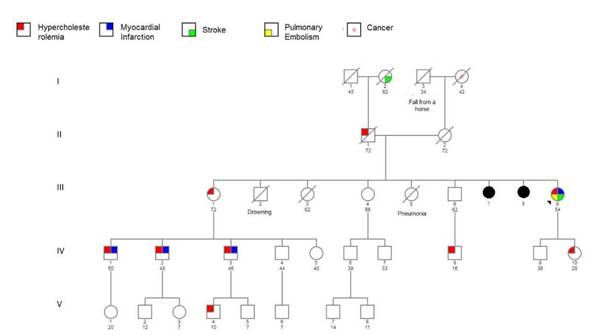
At 54 years old, the patient presented to the outpatient clinical genetics service, referred by her general practitioner whom suspected FH, at this visit the clinical geneticists constructed a genogram (figure 2) in which 10 individuals with hypercholesterolemia were identified, three of whom were diagnosed before 18 years. Premature
MI was documented in this woman <60 years (III9) and three men <55 years (IV1, IV2, and IV3). With this background, the Dutch Lipid Clinic Network (DLCN) criteria were applied, and the index case got 18 points (certainty >8 points).
Table 1 Case III9 Pharmacological Treatment.
| Medication | Dose | Indication |
|---|---|---|
| Rosuvastatin / Fenofibric acid | 10 mg/135 mg | Lipid-lowering, FH |
| Ahrocumab | 150 mg/day each 15 days | Lipid-lowering, FH |
| Aspirin | 100 mg/day | Antiplatelet, secondary prevention of CVD |
| Apixaban | 5 mg/day | Anticoagulant, secondary prevention of CVD |
| Sacubitril/Valsar-tan | 200 mg/day | Antihypertensive drugs with evidence in heart failure |
| Carvedilol | 25 mg/day | Beta-blocker, heart failure |
| Spironolactone | 25 mg/day | Aldosterone antagonist, heart failure |
| Esomeprazole | 40 mg/day | Proton pump inhibitor, gastric protection |
*FH: Familial Hypercholesterolemia. *CVD: Cardiovascular Disease.
Source: authors.
Genogram of the index case family is shown in figure 2. Despite that cases III9 and IV10 are the only ones with molecular confirmation of FH, many of the family members present hypercholesterolemia and have suffered from early ACVD. The first son (IV9) of the index case did not present elevated LDL cholesterol levels, nor major adverse cardiovascular events, not meeting Dutch criteria, therefore no molecular test was needed.
Consequently, at age 52, a molecular panel was carried out to identify the FH-related genes. Next-generation sequencing (NGS) analyzed LDLR, APOB, APOE, LDLRAP1, and PCSK9. A heterozygous pathogenic molecular variant associated with hypercholesterolemia, NM_000527.4:c.-135C>G, was found in LDLR promoter (Figure 3).
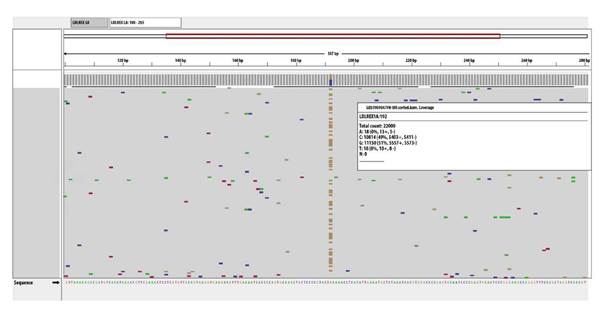
Source: authors.
Figure 3 Next-generation sequencing (NGS). NGS alignment of LDLR identifying the molecular pathogenic variant NM_000527.4:c.-135C>G in the gene promoter, which is frequently associated with hypercholesterolemia.
At age 52, with LDL-C levels of 325 mg/dL, alirocumab was added to rosuvastatin/fenofibric acid, obtaining an adequate response. In subsequent controls until this article's publication, LDL-C reduced to 210 mg/dL, ~34% less compared to previous values. Figure 4 shows the FH natural history and the lipid-lowering treatment in case III9.
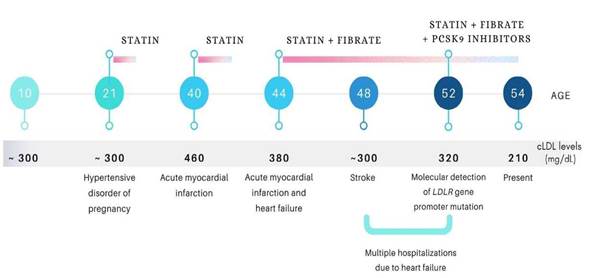
Source: authors.
Figure 4 Natural History of FH in Case III9. The series of medical events that have occurred throughout the index case's life, the LDL-C levels at different time points, and the lipid-lowering drugs administered are detailed chronologically. Note that the bar under the treatment sequence is interrupted when the patient discontinued pharmacological management for adverse effects.
Case two/IV10.
28-year-old female with FH, with no previous or ongoing pregnancies. At age 14, her LDL-C was tested due to family history. LDL-C was 290 mg/dL, but only healthy lifestyles were recommended. Since then, despite adequate non-pharmacological management, LDL-C (320 mg/dL) increased, so atorvastatin 40 mg/ day was initiated but with poor treatment adherence. Physical exam was unremarkable. At age 27, she went through NGS finding the same heterozygous variant, NM_000527.4:c.-135C>G in the LDLR promoter.
To date, after education on the FH consequences and the importance of self-care, she is under medical control, receives atorvastatin 40 mg/day + ezetimibe 20 mg/day, and goes through monthly LDL-C measurements. Her current LDL-C level is 102 mg/dL.
In cases III9 and IV10, a microdeletion involving the LDLR locus in the other allele was ruled out through MLPA, and, with that, the possibility that different abnormalities affected each allele.
Discussion
FH can be suspected when high levels of LDL-C are found, and is confirmed through predictive criteria. One should consider FH in patients <20 years with LDL-C >160 mg/dL or non-HDL cholesterol >190 mg/dL; in adults with LDL-C >190 mg/dL or non-HDL cholesterol >220 mg/dL; and in all individuals with premature ACVD. Some criteria allow making a possible, probable, or certain diagnosis of FH with or without a genetic test. The use of these tools has expanded because access to molecular confirmation is limited. Dutch Lipid Clinic Network (DLCN) criteria, the MEDPED criteria, and the Simon Broome Diagnostic Criteria from the United Kingdom9-12 evaluates personal and family history of hypercholesterolemia and premature ACVD, physical signs, LDL-C levels, and molecular tests, although a certainty score can be obtained without genetic testing. In the two cases described here, the DLCN criteria were applied; getting a score of 18 for case III9, 8 for case IV10, and 26 and 16 respectively once included in the molecular test.
The studies that were carried out in Colombia using molecular tests in individuals with FH, only in one case the variant c.-135C>G in the LDLR gene was identified7,8, this variant was the one described in the cases we report here, cases III9 and IV10. This variant is pathogenic according to ClinVar, Ensembl, and LDLR-LOVD databases. In ClinVar it was reported 10 times: Six times it was classified as probably pathogenic and four as pathogenic. The LDLR-LOVD database has five reports of this variant, in four appears as probably pathogenic and in one as pathogenic. In Ensembl, the variant is identified as rs879254375 and has two reported alleles c.-135C>G and c.-135C>A. All databases define it as a germline mutation associated with hypercholesterolemia that has an autosomal dominant inheritance pattern. In cases where molecular variants of the genes involved in the pathophysiology of FH are found, such variants must be studied using bioinformatic analyses to demonstrate their association with the disorder5,6,9.
FH integral management, which comprises healthy lifestyles and pharmacological therapy, corresponds to primary prevention when those affected do not present ACVD and secondary prevention when there is a personal history of ACVD (like cases IV10 and III9 respectively). LDL-C treatment goal depends on the presence of comorbidities (type 2 diabetes, chronic kidney disease, major adverse cardiovascular event) varying between <70 mg/dL and <160 mg/dL. Given the pathogenesis of the disease, therapeutic goals are rarely achieved despite adequate adherence to treatment with high intensity statins and patients often present therapeutic failure4,13,14.
Pharmacological management includes medications that can be used in monotherapy or combined. The first line are statins at the maximum tolerated dose. Statins inhibit HMG-CoA reductase, a limiting enzyme in the mevalonate pathway. As a result, there is augmented LDLR expression on the cell surface and greater mobilization of LDL-C to the intracellular compartment1,15,16. Like case III9, most FH patients don't achieve optimal LDL-C levels with statin monotherapy.
Ezetimibe plus statins is the next recommended step when monotherapy fails. Ezetimibe impairs cholesterol absorption by reversibly inactivating the intestinal transport protein NPC1L1. The IMPROVE- IT13 and the ENHANCE14,17 trials showed that dual therapy with ezetimibe and statins achieved a greater LDL-C reduction and improvement of cardiovascular outcomes compared to statin monotherapy.
Another strategy is adding PCSK9 inhibitors to statins. PCSK9 is involved in lysosomal degradation of LDLR. PCSK9 gain of function mutations explain the accumulation of LDL-C in the extracellular space. Evolocumab and alirocumab have been approved for patients who don't achieve LDL-C goals despite optimal statins monotherapy13,18-20. In the RUTHERFORD-2 study, which included HeFH patients with LDL-C >100 mg/dL on adequate lipid-lowering monotherapy, evolocumab reduced 60 % LDL-C levels at 12 weeks compared to placebo19. Case III9 achieved a 34 % reduction in LDL-C with rosuvastatin/ fenofibric acid plus alirocumab.
Lomitapide and mipomersen, were recently introduced for HoFH management. Lomitapide inhibits the microsomal triglyceride transfer protein (MTTP), which participates in the assembly and secretion of ApoB-containing lipoproteins. It has been approved to treat patients <18 years old, as it produces a dose-dependent reduction in cholesterol levels with an additive effect when co-administered with other lipid-lowering drugs20. Mipomersen is an antisense oligonucleotide that impairs the translation of Apo B100 mRNA. It is indicated for patients <12 years old20. These drugs have not yet demonstrated mitigation on ACVD risk, and are associated with hepatotoxicity, so they are included in the FDA's Risk Assessment and Mitigation Strategies program.
Conclusions
Early FH diagnosis and timely treatment can prevent ACVD. FH predictive criteria and genograms are useful tools to suspect FH and allow the identification of affected relatives, as well as a thorough history of present illness and physical exam. In families with hypercholesterolemia with an autosomal dominant or recessive inheritance pattern, measurement of LDL-C should be recommended to all members, and molecular tests performed to analyze the genes involved in FH. Multidisciplinary management is also required. Primary and secondary prevention should be provided to all patients with FH, as it aims to ensure the greatest number of ACVD-free years. The aim of this report is to show health providers how to assess hypercholesterolemia cases, even when the etiology is unknown.

