










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.18 no.3 Medellín July/Sept. 2005
ARTÍCULO DE REVISIÓN
Manejo de la insuficiencia adrenal
Approach to the handling of adrenal insufficiency
Danny Joan Orozco Arias1; Paula Andrea Pineda Bolívar1; Federico Uribe Londoño2
1. Estudiante de Medicina, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
2. Médico Internista y Endocrinólogo, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
RESUMEN
La insuficiencia adrenal (IA) se refiere a la hipofunción de dicha glándula debida a causas tanto primarias como secundarias, que resultan en niveles plasmáticos inadecuados de cortisol, andrógenos adrenales y adicionalmente, en la falla primaria de mineralocorticoides. Sus manifestaciones inespecíficas dificultan o retrasan con frecuencia el diagnóstico y tratamiento oportuno, lo cual incrementa la morbilidad y eventualmente la mortalidad de estos pacientes.
Se incluyen en este artículo: definición, epidemiología, causas, fisiopatología, clasificación, manifestaciones clínicas, diagnóstico y tratamiento de la IA. Además se consideran algunas situaciones especiales como la IA en el paciente críticamente enfermo y en la mujer gestante.
Finalmente se hace especial énfasis en un algoritmo diagnóstico, con la finalidad de facilitarle al médico general un enfoque ágil y oportuno de los pacientes con este problema endocrino.
PALABRAS CLAVE: cortisol, enfermedad de addison, insuficiencia adrenal
SUMMARY
The term adrenal insufficiency refers to the hypofunction of this gland. From the etiologic point of view it may be either primary or secondary. This insufficiency manifests as inadequate serum levels of cortisol and adrenal androgens in the secondary form and of these and mineralocorticoids in the primary one. Clinical manifestations are often nonspecific and, consequently, diagnosis may be difficult to establish and treatment may be delayed with increased morbidity and mortality.
This article on adrenal insufficiency includes its definition, epidemiology, etiology, physiopathology, classification, clinical presentation, diagnostic criteria and treatment guidelines. Besides, some special situations like critically ill patients and pregnant women are given special consideration. Emphasis is done on a diagnostic algorithm to make it easier for general practitioners the approach to patients with this endocrine disorder.
KEY WORDS: addison's disease, adrenal insufficiency, cortisol
DEFINICIÓN
El término IA se refiere a la hipofunción de esta glándula debida a diversas causas, y que da lugar a una sintomatología muchas veces inespecífica, que puede ser permanente o transitoria dependiendo de los eventos estresantes a los que la persona esté sometida y de la enfermedad causal. De acuerdo con el sitio afectado del eje hipotálamo–hipófisisadrenales se la denomina primaria si la alteración es en las glándulas adrenales (baja producción de cortisol y alta concentración plasmática de hormona adrenocorticotrópica, ACTH); en cambio, se la denomina secundaria si la alteración ocurre en el hipotálamo o en la hipófisis (niveles bajos de hormona liberadora de corticotropina, CRH, y/o ACTH y cortisol).1,2
EPIDEMIOLOGÍA
La incidencia de la IA primaria es de 4.7–6.2 casos por cada millón de adultos por año, con una prevalencia de 93–140 por cada millón de personas.1
Aparece más comúnmente en la cuarta década de la vida, y afecta con mayor frecuencia a las mujeres.1
La IA secundaria tiene una prevalencia estimada de 150–280 casos por millón de personas. Al igual que la IA primaria, afecta más frecuentemente a las mujeres que a los hombres; la edad promedio en el momento del diagnóstico es de 50 años.1
ETIOLOGÍA
Las causas de la IA son múltiples; en la actualidad predomina la forma primaria autoinmune en tanto que ha disminuido la frecuencia de las debidas a hemorragias (tratamientos con warfarina o heparina, síndrome antifosfolípido y síndrome de Waterhouse–Friderichsen por meningococcemia) o a infecciones, principalmente granulomatosas, como la tuberculosis; la disminución se debe probablemente a los avances médicos, entre ellos, el control de los microorganismos. La IA primaria autoinmune puede presentarse de manera aislada (enfermedad de Addison) o haciendo parte del síndrome poliglandular autoinmune (SPA), en el que está comprometida la función de varias glándulas, conformando cuadros clínicos conocidos como SPA tipo I y SPA tipo II.2 En el tipo I, la IA se acompaña de hipoparatiroidismo y candidiasis mucocutánea crónica. En el tipo II, la IA se asocia a tiroiditis autoinmune, diabetes mellitus tipo 1 e insuficiencia ovárica prematura.2 Otras causas menos comunes de IA primaria se resumen en la tabla Nº 1.
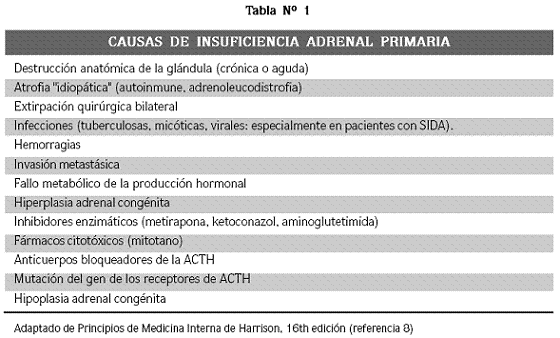
En la IA secundaria las alteraciones hipotálamohipofisiarias llevan a la ausencia de estimulación adrenal por la hormona adrenocorticotrófica –ACTH– lo cual conduce a la atrofia adrenal (capas fasciculada y reticular, ya que la capa glomerulosa depende fundamentalmente del eje reninaangiotensina–aldosterona). La terapia glucocorticoide se ha convertido en el principal factor desencadenante de IA secundaria luego de un tratamiento por más de dos semanas seguido de supresión súbita del medicamento; la magnitud y duración de la atrofia adrenal secundaria dependen directamente de la dosis y del tiempo de administración.1,3 También se observa IA secundaria en pacientes con lesiones quirúrgicas o por irradiación de las células corticotropas luego del tratamiento de tumores hipofisiarios primarios o metastáticos. Otros procesos destructivos, poco frecuentes, en la región de la silla turca como la sarcoidosis, las infecciones (tuberculosis, actinomicosis, nocardiosis) y los accidentes vasculares como el síndrome de Sheehan, pueden conducir igualmente a una insuficiente producción de ACTH.3–5 Estas y otras causas de IA secundaria se presentan en la tabla Nº 2.

FISIOPATOLOGÍA
La destrucción progresiva de la glándula que caracteriza la IA primaria de carácter autoinmune (enfermedad de Adisson) se lleva a cabo por la presencia en el 50% de los pacientes de anticuerpos específicos contra la 17Αhidroxilasa y la 21α hidroxilasa,2,6 enzimas citosólicas que intervienen en la síntesis hormonal. Sin embargo, parece que es más relevante en el daño glandular, la acción de los linfocitos T citotóxicos que conduce a las manifestaciones clínicas solo cuando la pérdida del tejido adrenal alcanza un 80 a 90%.2 Esto ocasiona incapacidad para la síntesis y liberación de los productos hormonales de la glándula, con lo que disminuyen las concentraciones plasmáticas de cortisol, aldosterona y dehidroepiandrosterona (DHEA) y aumenta la ACTH, por el mecanismo de retroalimentación (feedback). La situación es diferente en la IA secundaria en la cual solo se presenta deficiencia de cortisol y DHEA, debido a que en esta enfermedad es normal la secreción de mineralocorticoides pues su regulación depende del sistema renina–angiotensina que es independiente del hipotálamo y la hipófisis.1
En la IA secundaria desencadenada por la supresión de glucocorticoides, el mecanismo de retroalimentación negativa ocasionada por los altos niveles de estas hormonas durante la terapia, conduce a una reducción del estímulo endógeno de las adrenales con la consiguiente atrofia glandular (tanto hipofisiaria como adrenal), lo que lleva a que al suspender los glucocorticoides sea inadecuada la producción endógena de cortisol.3,4 El déficit o el incremento de las hormonas propias del eje hipotálamo–hipófisis–adrenal (HHA) explican algunas diferencias en los signos y síntomas que caracterizan las diferentes formas de presentación de la enfermedad; tener esto en cuenta orienta al clínico en la elección de los exámenes paraclínicos necesarios para confirmar el diagnóstico.
CLASIFICACIÓN
Los pacientes con IA se pueden dividir en dos grandes grupos:
• Los debidos a incapacidad primaria de la glándula para elaborar hormonas en cantidad suficiente.
• Los secundarios a la producción o liberación insuficiente de ACTH.
Otra manera de clasificar la IA es tener en cuenta la evolución del cuadro clínico que puede ser agudo o crónico. Esto es de valor para identificar el posible factor causal. Así, por ejemplo, la IA aguda se debe frecuentemente a causas infecciosas, hemorrágicas y a la suspensión súbita de los glucocorticoides. En contraste, la IA crónica obedece más a procesos autoinmunes o granulomatosos que van alterando progresivamente la glándula y se hace manifiesta inicialmente solo en determinadas situaciones.
MANIFESTACIONES CLÍNICAS
Los síntomas de la IA crónica se caracterizan por un comienzo insidioso, debido a la destrucción lenta y progresiva de la glándula. En cambio, la IA aguda puede presentarse como un choque fulminante tal como el que describieron Waterhouse y Friderichsen. Es importante tener en cuenta que una destrucción progresiva de la glándula puede manifestarse solo en determinados estados de estrés que permiten evidenciar la falta de reserva adrenal, lo que se convierte en el primer indicio de la insuficiencia hormonal en el curso de procesos agudos que actúan como eventos desencadenantes.1,3,4
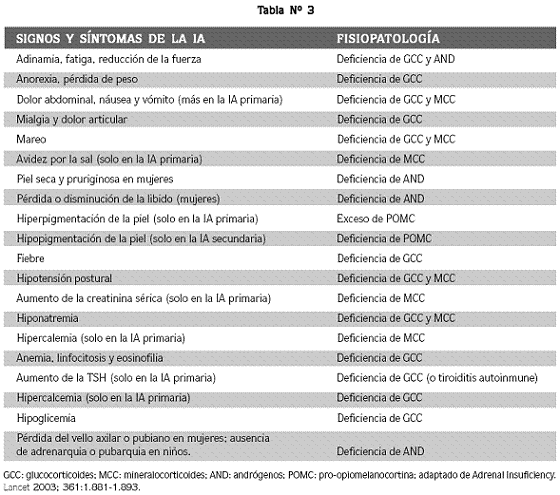
Los signos y síntomas de la IA pueden ser muy inespecíficos, pero la sospecha clínica es fundamental en todo paciente con astenia, pérdida de peso, anorexia, náusea y vómito. La hiperpigmentación cutánea y la hipotensión postural pueden o no acompañar el cuadro y, en caso de presentarse, sugieren destrucción glandular primaria. La sintomatología de la IA se corresponde con una fisiopatología hormonal específica como se muestra en la tabla Nº 3.
DIAGNÓSTICO
Luego de una historia clínica cuidadosa y un examen físico minucioso, que conduzcan a la sospecha de IA, se debe proseguir con la evaluación bioquímica de la función adrenal; para ello se recurre a los siguientes exámenes:
Secreción basal de cortisol
En un paciente ambulatorio y sin una enfermedad sistémica importante asociada, el primer parámetro de laboratorio que debe evaluarse es la concentración de cortisol sérico a las 8 a.m. Valores por debajo de 5µg/dL son diagnósticos de la enfermedad. Entre 5 y 10 µg/dL son sugestivos pero no permiten hacer el diagnóstico y un valor de 18 µg/dL o mayor descarta la presencia de esta enfermedad.3 Al interpretar los resultados debe tenerse en cuenta que los pacientes con IA parcial pueden tener niveles basales de cortisol dentro del rango normal pero una respuesta inadecuada al estrés; para aclarar el diagnóstico es muy importante una prueba dinámica de estimulación con ACTH.
Prueba de estimulación con ACTH
Esta prueba es el estándar de oro debido a que permite evaluar la capacidad de reserva funcional de la glándula para producir cortisol y a que se puede realizar a cualquier hora del día; además, es rápida, estable, no interfiere con la dieta ni con la alimentación, es fácil de interpretar y se puede aplicar a todas las personas sin efectos colaterales importantes. Para hacerla se administran 250 µg de cosyntropin (Cortrosyn® o Synacthen® –ACTH sintética–) por vía intramuscular o intravenosa y se miden los niveles de cortisol antes de su aplicación, 30 y 60 minutos después. En condiciones normales el valor de cortisol a los 60 minutos debe sobrepasar los 18 ó 20 µg/dL, según diferentes autores.1–7 Valores inferiores a estos indican grados diversos de falla adrenal o baja reserva.
Concentración plasmática de ACTH
La prueba de estimulación con ACTH hace el diagnóstico de la IA pero para diferenciar si la alteración es primaria o secundaria, se requiere medir la concentración plasmática de ACTH a las 8 de la mañana; está elevada en la forma primaria y disminuida en la secundaria. La cifra aceptada como normal es 6–76 pg/mL (o 1.3–16.7 pmol/L).2 A pesar de que los signos y síntomas pueden hacer pensar fuertemente en una u otra causa, es necesario llevar a cabo esta prueba para confirmar el tipo de falla adrenal y así poder emprender el tratamiento.3,6
Ayudas complementarias
Además de estas pruebas, que son específicas para detectar la anomalía endocrina, existen otras que miden indirectamente el comportamiento hormonal y orientan sobre las posibles causas.
En la IA primaria los líquidos y electrólitos están alterados debido a la deficiencia de mineralocorticoides, lo que se evidencia por hiponatremia, hipercalemia y deshidratación. El hemoleucograma ayuda a corroborar las causas infecciosas y a veces se encuentra eosinofilia moderada. La deshidrogenasa láctica es una prueba a favor del diagnóstico de metástasis tumoral cuando esta se sospecha.
La medición de anticuerpos no se utiliza de manera rutinaria debido a su baja sensibilidad ya que solo se detectan en el 50% de los pacientes con IA primaria.5
Otras pruebas para evaluar la respuesta del eje hipófisis–adrenal como la de tolerancia a la insulina y la de la metirapona no se llevan a cabo de manera rutinaria sino solo en pacientes seleccionados, como se muestra en el flujograma de la figura Nº 1.7

Cuando está firmemente establecido el nivel de la falla adrenal se requiere complementar la evaluación del paciente con estudios imaginológicos (TAC o RM) de las adrenales en caso de IA primaria y de la hipófisis en caso de IA secundaria.
TRATAMIENTO
Principios generales
Todos los pacientes con IA deben ser tratados con hormonoterapia sustitutiva que corrija los déficit de glucocorticoides y mineralocorticoides.1,3,–6 Es muy importante la educación que se brinda al paciente y a su familia sobre los siguientes aspectos de esta enfermedad:
• La naturaleza del déficit hormonal y el uso racional de la terapia de reemplazo.
• El mantenimiento de la medicación.
• El cambio en la dosis de la medicación durante enfermedades intercurrentes no urgentes.
• Cuándo consultar al médico.
• Cuándo y cómo inyectar glucocorticoides en las emergencias.
• Sistema de identificación para el manejo en urgencias (credencial o escarapela).3
En los países desarrollados1,3 las placas o el carné de identificación han sido una medida eficaz y oportuna para el tratamiento en las situaciones de emergencia de estos pacientes.
La terapia farmacológica se basa en la administración de hidrocortisona 20–30 mg/día o su equivalente; para imitar el ritmo adrenal diurno se toman dos tercios de la dosis total en la mañana y un tercio al final de la tarde. Se recomienda que debido a los efectos secundarios gastrointestinales de los glucocorticoides se los tome con las comidas, leche o antiácidos.1,2,4
Algunos autores plantean que sería mejor llevar a cabo el reemplazo hormonal con dexametasona debido a que su vida media es más prolongada y mantendría la ACTH en un nivel normal en las horas de la noche, sin permitir las fluctuaciones en la concentración plasmática que se observarían entre las dosis con los esteroides de vida media corta como la hidrocortisona.3 Sin embargo, hay más tendencia a la utilización de esta última porque simula mejor las variaciones en las concentraciones fisiológicas cuando se la administra en varias dosis diarias.1,4–8 En Colombia no se dispone por el momento de la forma oral de hidrocortisona lo cual hace necesario recurrir a dosis equivalentes y fraccionadas de otros glucocorticoides como la prednisolona o la prednisona (5–7.5 mg/día).
Cuando la IA es primaria, siempre es necesario el reemplazo de mineralocorticoides que se hace mediante la administración de fludrocortisona 0.05–0.1 mg/día por vía oral y adicionalmente se debe aconsejar a los pacientes que tomen constantemente una cantidad generosa de sal (3–4 g/día). En situaciones de máximo estrés se hace necesaria la administración IV de hidrocortisona; si la dosis es mayor de 100 mg/día no es necesario agregar fludrocortisona, ya que a estas dosis la hidrocortisona tiene actividad mineralocorticoide.1,4–8
Es importante hacer un control constante para evaluar el exceso o el déficit hormonal. Se recomienda vigilar la aparición de los estigmas cushinoides que hablan de una sobredosificación de glucocorticoides. La reaparición de la sintomatología inicial y la hipotensión postural indican una subdosificación que también es evidente por hallazgos en los exámenes de laboratorio como hiponatremia e hipercalemia.4
El reemplazo de andrógenos en las mujeres con IA ha mostrado efectos positivos en su bienestar, estado de ánimo y densidad ósea, por lo que se recomienda administrarles DHEA a dosis de 25–50 mg al día. El déficit de andrógenos suprarrenales en los hombres no es notable ni hay que reemplazárselos ya que la principal fuente de estos en el sexo masculino son las gónadas.1,7–9
En la insuficiencia adrenal secundaria no se presenta déficit de mineralocorticoides debido a que la producción de estos en la capa glomerulosa adrenal depende del sistema renina–angiotensina y no de la concentración plasmática de ACTH. Sin embargo, es necesario tener en cuenta que con frecuencia coexiste el déficit de ACTH con otras deficiencias hormonales de la adeno–hipófisis que requieren tratamiento, como hipotiroidismo y/o hipogonadismo secundarios. Cuando hay deficiencia de hormona del crecimiento puede haber una mayor propensión a la hipoglicemia y en los niños o adolescentes que no han terminado su crecimiento debe hacerse reemplazo. En los adultos puede llegar a justificarse dicho reemplazo si persisten los signos o síntomas de su deficiencia (debilidad, atrofia muscular, etc.) y después de haber reemplazado los glucocorticoides, la levotiroxina y los esteroides gonadales (dependiendo de las deficiencias asociadas). Si coexisten la insuficiencia adrenal y el hipotiroidismo es importante iniciar primero el reemplazo adrenal y luego el tiroideo puesto que, de hacerlo en orden inverso, el aumento en la actividad metabólica con el suministro de la hormona tiroidea puede desencadenar una crisis adrenal aguda en el paciente con IA todavía no reemplazada. Cuando la IA secundaria se debe a una supresión prolongada del eje hipotálamo–hipófisis–adrenal, como en el caso de un tratamiento prolongado con dosis suprafisiológicas de glucocorticoides o en pacientes con hipercortisolismo endógeno ‘curados' quirúrgicamente, la falla adrenal puede durar hasta 6 ó 12 meses, período durante el cual se debe suplementar al paciente con dosis fisiológicas de glucocorticoides hasta la recuperación del eje. Con respecto al reemplazo con glucocorticoides, las dosis y otras recomendaciones, se aplican los mismos parámetros en la IA primaria y en la secundaria.
Insuficiencia adrenal de novo
Para los pacientes en quienes se sospecha IA y la sintomatología amerita el tratamiento de urgencia, algunos autores recomiendan seguir un esquema que permita no solo remediar la situación aguda con el reemplazo hormonal sino también hacer el diagnóstico bioquímico de la IA. Debido a esto, la dexametasona es una excelente alternativa ya que la reposición inmediata con ella de la deficiencia gucocorticoide no interfiere en las mediciones séricas o urinarias de cortisol como sí pasa con otros glucocorticoides.3 Sin embargo, se debe pasar al manejo con hidrocortisona una vez llevadas a cabo las evaluaciones bioquímicas necesarias del funcionamiento glandular. El tratamiento del episodio agudo se basa en la reposición del volumen sanguíneo con soluciones que no sean hipotónicas para no empeorar la hiponatremia existente. La fludrocortisona solo se debe administrar una vez que el paciente haya iniciado la ingesta alimentaria e hídrica;3 no tiene fundamento hacerlo inmediatamente, debido a que su acción tarda en el tiempo y la reposición del volumen y de los electrólitos puede hacerse de manera rápida y eficaz mediante soluciones parenterales.
Enfermedades intercurrentes
Durante las enfermedades intercurrentes es necesario, en especial si hay fiebre, duplicar la dosis de hidrocortisona o del glucocorticoide con el cual se esté haciendo el reemplazo.1,8 Si la enfermedad es grave, deben darse 75–150 mg/día de hidrocortisona. Del mismo modo, deben administrarse glucocorticoides adicionales antes de una intervención quirúrgica mayor y en los casos de trauma y de enfermedades que requieran monitorización en cuidados intensivos, por infusión intravenosa de 100–150 mg de hidrocortisona en dextrosa al 5% en 24 horas.1,8 También está indicado el incremento de la dosis de fludrocortisona y de la sal en la dieta cuando se hace ejercicio físico intenso o se suda mucho;1,2 si se tienen trastornos gastrointestinales como diarrea o vómito, algunos autores recomiendan administrar glucocorticoides por vía parenteral y ajustar las dosis dependiendo del curso de la enfermedad.1,3
En casos de estrés intenso donde el incremento de la dosis fue importante y por varios días, la disminución del tratamiento glucocorticoide debe hacerse progresivamente hasta alcanzar los valores de mantenimiento necesarios para permanecer libre de sintomatología en la mejor condición posible.1,3,6
Situaciones especiales
Pacientes críticamente enfermos
Los pacientes críticamente enfermos ameritan una atención especial debido a que en ellos el eje hipotálamo–hipófisis–adrenal está muy activado. La gran cantidad de citoquinas liberadas en ellos tiene hasta cierto punto un papel protector ya que se estimulan la activación del eje y la afinidad de los glucocorticoides por su receptor.10
Sin embargo, en pacientes sépticos, la liberación masiva de mediadores inflamatorios puede interferir directamente con la síntesis de cortisol por las glándulas adrenales11 e inducir resistencia periférica, contrario a lo que una respuesta normal ocasionaría.12 Esto puede llevar a que la respuesta normal sea insuficiente.
No solo la condición crítica de los pacientes sino también todo el manejo farmacológico que se hace, interfieren con el adecuado funcionamiento adrenal. La acción de fármacos utilizados en unidades de cuidado intensivo como el anestésico etomidato o antifúngicos como el ketoconazol son ejemplos de alteración en la actividad enzimática en la síntesis de cortisol.13
Desafortunadamente, no existe consenso acerca de cómo diagnosticar la IA en pacientes críticamente enfermos, pero se tiene claro que los niveles de cortisol para responder adecuadamente a situaciones de estrés deben ser más altos que los basales. Incluso se dice que deben ser de 30µg/dL8 o más y que en caso de encontrar una cifra menor se debe sospechar una IA primaria o secundaria. Las crisis adrenales pueden presentarse en algunos pacientes críticamente enfermos a pesar de tener niveles basales de cortisol dentro del rango normal, pero con respuesta deficiente al estímulo con ACTH.13
Otros autores han propuesto que para los pacientes críticamente enfermos se puede considerar normal en una muestra aleatoria de cortisol plasmático un valor superior a 34 µg/dl.13 Si el resultado es menor de 15 µg/dL se considera segura la IA, y si el valor está entre 15 y 33 µg/dL es muy probable que esté presente la IA y que el paciente se beneficie de la terapia con glucocorticoides. Sin embargo, en este rango debe confirmarse el diagnóstico por medio de una prueba de estimulación con ACTH. Si el incremento sobre el valor basal a los 30 ó 60 minutos es superior a 9 µg/dL se considera improbable la IA. En caso de que la prueba dinámica descarte la IA se debe desmontar la terapia de reposición.13
Algunos autores sostienen que la liberación de citoquinas en pacientes críticamente enfermos podría dañar las células hipofisiarias corticotropas con la consiguiente disminución en la producción de ACTH y producir una IA secundaria.1 Por esto se recomienda hacer medición de cortisol y ACTH plasmáticos en los pacientes críticamente enfermos con sospecha de IA, seguida de la administración de hidrocortisona a razón de 50 mg IV cada 6 horas, la cual debe ser desmontada si el nivel de cortisol descarta la IA que se sospechaba,1,13 como antes se indicó.
Gestantes
Antes de que estuviera disponible la terapia de reposición hormonal con glucocorticoides y mineralocorticoides, la mortalidad de las mujeres gestantes con IA se calculaba entre 35–45%. Ahora las pacientes con IA pueden sobrellevar su gestación sin mayores dificultades.3 Las dosis de glucocorticoides y mineralocorticoides deben continuarse durante toda la gestación. Algunas mujeres requieren un incremento leve en la dosis de glucocorticoides durante el tercer trimestre.3 Sin embargo, otros autores recomiendan aumentarla en un 50% durante el último trimestre.1
Durante el trabajo de parto se recomiendan una adecuada hidratación con solución salina y 25 mg de hidrocortisona intravenosa cada 6 horas.3 La dosis se puede incrementar hasta 100 mg cada 6 horas si el parto se prolonga demasiado.1 Luego puede ser disminuida progresivamente para alcanzar los niveles de mantenimiento en 3 días.3
Comorbilidades
Otra situación que debe tenerse en cuenta en los pacientes que reciben la terapia de reemplazo es el incremento de la dosis cuando se utilizan concomitantemente otros medicamentos que aceleran el metabolismo hepático como la fenitoína, los barbitúricos, la rifampicina, el mitotano y la aminoglutetimida.3,6
CONCLUSIONES
Dada la inespecificidad de la mayoría de los signos y síntomas de la IA es muy importante la sospecha clínica para poder establecer un diagnóstico correcto y dar un tratamiento adecuado en estos pacientes. El laboratorio clínico moderno ofrece un gran apoyo en el diagnóstico preciso de esta enfermedad. Los retrasos en su diagnóstico producen alteraciones significativas en la calidad de vida y ponen al paciente en riesgo de muerte principalmente cuando hay enfermedades asociadas o situaciones críticas. La supervivencia a largo plazo de los pacientes con IA se basa en gran medida en prevenir y tratar las crisis adrenales.
REFERENCIAS BIBLIOGRÁFICAS
1. WIEBKE B. Adrenal Insufficiency. Lancet 2003; 361: 1.881–1.893. [ Links ]
2. BETTERLE C, DAL PRA C, MANTERO F, ZANCHETTA R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens and their applicability in diagnosis and disease prediction. Endocr Rev 2002; 23: 327–364. [ Links ]
3. STEWART PM. The Adrenal Cortex. In: Larsen PR, et al, eds. William's Texbook of Endocrinology, 10th ed. Philadelphia: Saunders; 2003. [ Links ]
4. LORIAUX DL, MC DONALD WJ. Adrenal insufficiency. In: DeGroot LJ, et al, eds. Endocrinology, 4thed. Philadelphia: Saunders; 2001. [ Links ]
5. ORREGO JJ. Adrenales. En: Orrego A, ed. Fundamentos de Medicina: Endocrinología, 6ª ed. Medellín: CIB; 2004. [ Links ]
6. FITZGERALD PA. Hypothalamic and pituitary hormones. In: Katzung B, ed. Basic Clinical Pharmacology, 9th ed. Estados Unidos. McGraw Hill–Interamericana; 2003. [ Links ]
7. DORIN R, QUALLS C, CRAPO L. Diagnosis of adrenal insufficiency. Ann Intern Med 2003; 139: 194–204. [ Links ]
8. WILLIAMS GH, DLUHY RG. Adrenal cortex diseases. In: Braunwald E, et al, eds. Harrison's Principles of Internal Medicine, 16th ed. Estados Unidos. McGraw Hill–Interamericana, 2004. [ Links ]
9. HUNT PJ, GURNELL EM, HUPPERT FA, et al. Improvement in mood and fatigue after dehydroepiandrosterone replacement in Addison's disease in a randomized, double blind trial. J Clin Endocrinol Metab 2000; 85: 4.650–4.656. [ Links ]
10. FRANCHIMONT D, MARTENS H, HAGELSTEIN MT, et al. Tumor necrosis factor alpha decreases, and interleukin–10 increases, the sensitivity of human monocytes to dexamethasone: potencial regulation of the glucocorticoid receptor. J Clin Endocrinol Metab 1999; 84: 2.834–2.839. [ Links ]
11. WAGNER RL, WHITE PF, KAN PB, ROSENTHAL MH, FELDEMAN D. Inhibition of adrenal steroidogenesis by the anesthetic etomidate. N Engl J Med 1984; 310: 1.415–1.421. [ Links ]
12. MOLIJN GJ, SPEK JJ, VAN UFFELEN JC, et al. Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. J Clin Endocrinol Metab 1995; 80: 1.799–1.803. [ Links ]
13. COOPER MS, STEWART PM. Corticosteroid insufficiency in acutely ill patiens. N Engl J Med 2003; 348: 727–734. [ Links ]
Recibido: 02 de febrero de 2005
Aceptado: 21 de julio de 2005

