











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
La linfohistiocitosis hemofagocítica (LHH), más conocida en la literatura médica como síndrome hemofagocítico, es caracterizada por una activación excesiva de naturaleza no maligna de los macrófagos y los linfocitos T citotóxicos (LTc), como resultado de una citotoxicidad defectuosa asociada a una marcada hipercitoquinemia 1. Fue descrita por primera vez en 1939, pero su caracterización se logró en 1979 y desde entonces se han realizado múltiples modificaciones en sus criterios diagnósticos 2.
La LHH se clasifica en primaria y secundaria, de acuerdo con la presencia o no de defectos genéticos, respectivamente (Tabla 1). La LHH primaria a su vez se subdivide en linfohistiocitosis hemofagocítica familiar con herencia autosómica recesiva, causada por mutaciones en los genes implicados en la expresión de las proteínas de tráfico de los gránulos citotóxicos 3; LHH asociada a síndromes genéticos (Chediak Higashi, Griselli y Hermansky Pudlak) y la enfermedad linfoproliferativa asociada al cromosoma X. Usualmente las manifestaciones clínicas de las LHH primarias se presentan en los primeros 12 meses de vida, con una supervivencia promedio aproximada de 10 años 2,4,5. Las formas secundarias o adquiridas se manifiestan principalmente en la población adulta, pero pueden manifestarse en la población infantil 6-13, presentándose frecuentemente en el contexto de las infecciones, tumores malignos y enfermedades autoinmunes.
Adicionalmente, se han reportado algunos casos de LHH en pacientes con trasplante de órgano sólido bajo terapia inmunosupresora, con una incidencia reportada de 0,4-0,5 % 14, y con una mortalidad del 80 %, especialmente cuando se asocia con una falla multisistémica 2,4,15,16. Karras y col. observaron que la presencia de organomegalia, elevación de las aminotransferasas, prolongación del tiempo de protrombina y trombocitopenia, fueron factores de riesgo de muerte en pacientes con LHH posterior a un trasplante renal 15.
Tabla 1 Clasificación de la linfohistiocitosis hemofagocítica
| LHH PRIMARIA | LHH SECUNDARIA |
|---|---|
| Linfohistiocitosis hemofagocítica familiar tipo 1 al 5 Asociada a síndromes genéticos: síndrome de Chediak Higashi síndrome de Grisselli síndrome de Hermansky Pudlak Enfermedad linfoproliferativa asociado al cromosoma X | Infecciones Cáncer Medicamentos Desórdenes autoinmunes, autoinflamatorios |
Fuente: 3.
En la actualidad existe poca literatura con respecto a la LHH en pacientes trasplantados de riñón, siendo en su mayoría proveniente de reportes o series de casos (Tabla 2) 11,12,14,17-20.
El poco conocimiento de la LHH postrasplante, sumado a su presentación clínica inespecífica, hace que su diagnóstico sea un reto. Esta revisión pretende evaluar el estado de arte actual de la LHH postrasplante renal, permitiendo al clínico adquirir un conocimiento actualizado sobre la fisiopatología, evolución clínica, el diagnóstico y tratamiento de esta entidad.
Tabla 2 Reporte de casos de LHH en trasplante renal
| Series de casos | ||||||||
|---|---|---|---|---|---|---|---|---|
| Año (ref) | n.o pacientes | Causa n.° (%)* | Reducción Inmunosupresión | Rechazo | Qx | Muerte | Pérdida del injerto | |
| 1979 (21) | 19 | 14 (73,7 %) | 14 (73,7 %) | Sí | No | No | Sí | |
| 2004 (15) | 17 | 13 (76,5 %) | 16 (94,1 %) | Sí | No | No | Sí | |
| Reportes de casos | ||||||||
| Año (ref) | Días postrasplante | Condición asociada | Inducción depletores linfocitos | Reducción inmunosupresión | Qx | Muerte | Pérdida del injerto | |
| 2016 (11) | 5 | CMV | Sí | No | No | Sí | - | |
| 2013 (14) | 36 | Rechazo agudo | Sí | Sí | No | No | No | |
| 2014 (12) | 300 | BK | Sí | Sí | No | No | Sí | |
| 2006 (18) | 10 | Toxoplasm | Sí | Sí | No | Sí | - | |
| 2008 (19) | 700 | PVB19 | No descrito | Sí | No | No | No | |
Qx: quimioterapia. *: causa identificada que desencadenó la LHH. CMV: infección por citomegalovirus. BK: nefropatía por BK virus. Toxoplasm: toxoplasmosis. PVB19: parvovirus B19. Fuente: 11,12,14,15,18,19,21.
FACTORES DE RIESGO
Las formas secundarias de la LHH están relacionadas con una respuesta inmune alterada, lo cual es una característica de los pacientes inmunosuprimidos, bien sea por infecciones o por tratamiento para enfermedades autoinmunes, neoplásicas o trasplante (Tabla 1) 6-12. En los pacientes trasplantados esta condición se puede presentar especialmente en las primeras semanas postrasplante y se relaciona frecuentemente con infecciones, la presencia de malignidades y el uso de medicamentos depletores de linfocitos 22.
Existe una relación compleja entre los procesos infecciosos y la LHH, esto debido a las infecciones que pueden causar este síndrome en personas predispuestas genéticamente o a la leucopenia secundaria a la LHH que puede favorecer el desarrollo de un proceso infeccioso. Los microorganismos más frecuentemente asociados son los virus, especialmente los herpesvirus, por infección primaria o reactivación. También se han descrito infecciones bacterianas, micobacterianas, parasitarias y fúngicas (Tabla 3) 2. En relación a las infecciones fúngicas, los hongos del género histoplasma son los causantes más frecuentes de LHH en los pacientes inmunosuprimidos por el virus de la inmunodeficiencia humana (VIH) y por el trasplante renal 8-10; hallazgo a tener en cuenta ya que la histoplasmosis a su vez se presenta en forma no despreciable en los pacientes trasplantados de órgano sólido, un estudio realizado en la ciudad de Medellín reportó una incidencia del 1,1 % en los pacientes trasplantados renales 9.
Tabla 3 Causas infecciosas del síndrome hemofagocítico
| Virus | Bacterias | Parásitos y hongos |
|---|---|---|
| Herpesvirus: virus herpes simplex, varicela-zóster, virus de Epstein-Barr, citomegalovirus, herpesvirus 6, herpesvirus 8. Parvovirus B19 Adenovirus Enterovirus VIH Polyomavirus Arbovirus Virus Coxsackie | Micobacterias Salmonella Borrelia burgdorferi Leptospira Brucella tularensis Chlamydia psittaci Mycoplasma pneumoniae Ehrlichia Rickettsias Legionella | Babesia Leishmania Toxoplasma gondii Plasmodium Pneumocystis jirovecii Candida Aspergillus Cryptococcus Histoplasma |
Fuente: 2.
De forma similar, existe una relación circular entre los procesos malignos y la LHH. Esta asociación es mayor con los tumores malignos linfoproliferativos (Linfoma de células T y B), específicamente en los inducidos por virus, principalmente el virus Epstein-Barr (EBV), siendo uno de los más frecuentes en la enfermedad linfoproliferativa asociada al trasplante (PTLD, del inglés Posttransplant lymphoproliferative disease) 23-25. Con menos frecuencia se asocia a tumores malignos sólidos 26,27, los cuales también se pueden presentar luego del trasplante. En las enfermedades autoinmunes (artritis crónica juvenil, enfermedad de Still, lupus eritematoso sistémico, esclerosis sistémica, artritis reumatoide), la LHH se puede presentar espontáneamente o relacionada con la terapia inmunosupresora y las infecciones. Por otro lado, en los últimos años han incrementado los reportes de casos que asocian esta entidad con la administración de biológicos (nivolumap, ipilimumab, etanercept, prembrolizumab) 28, fármacos convencionales (vancomicina, trimetropim-sulfametoxazol, fenobarbital, lamotrigina, fenitoina, metrotexate, sulfasalazina, ácido retinoico) y nutrición parenteral, terapias que son utilizadas frecuentemente en los pacientes trasplantados 6,29.
FISIOPATOLOGÍA
En condiciones normales ante una infección viral o en la presencia de células tumorales, las células que presentan antígenos activan los linfocitos CD8+ citotóxicos (LTc) y las células Natural Killer (NK), las cuales eliminan las células infectadas (o células tumorales) por medio de la lisis mediada por perforinas (Figura 1a). Los LTc responden a la activación de sus receptores por patrones moleculares asociados a daño (DAMPS) o a patógenos (PAMPS). Posterior a su activación, estas células producen a nivel intracelular granzimas (Gz) y perforinas (Perf) por el aparato de Golgi. Una vez formado el gránulo citotóxico, este migra a la membrana celular por la señalización ofrecida por las proteínas en la pared del gránulo, entre ellas las proteínas munc 13-4, munc 18-2 y la sintaxina 11 que favorecen su adhesión y posterior liberación en la sinapsis inmunológica (Figura 1b) 30.
Aunque la forma de acción reconocida para la citotoxidad corresponde a la formación de poros en las células diana (células presentadoras de antígenos) por las Perf y posterior penetración de las Gz; en años recientes se han identificado procesos de endocitosis de las Gz y Perf, con el posterior anclaje de las Perf en la pared de los endosomas y la liberación intracelular de las Gz. Estas últimas interactúan y clivan los dominios de los agonistas de muerte (BID: BH3 interacting-domain death agonist), que a su vez permiten la permeabilización de la membrana externa mitocondrial para la activación de citocromos y caspasas, que inducen apoptosis celular (Figura 1b).
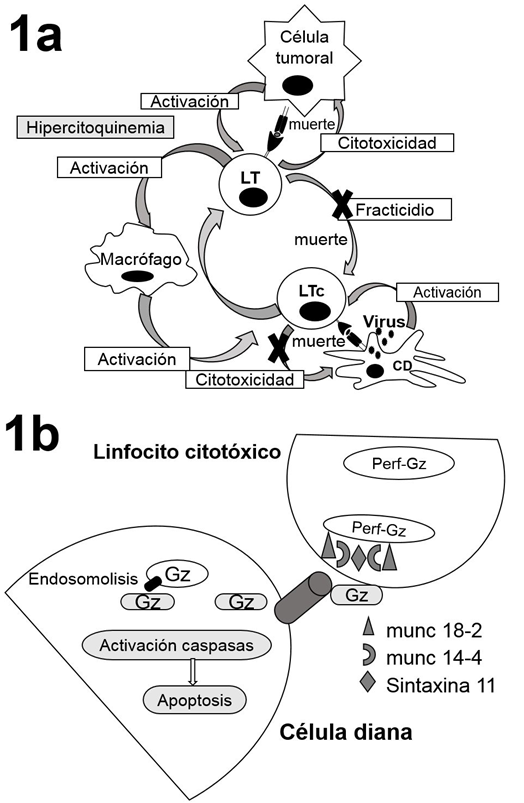
Figura 1 Fisiopatología de la LHH. En la figura 1a se muestra la activación de los linfocitos T citotóxicos (LTc) por las células dendríticas, la cual lleva a la lisis celular mediada por perforinas. Estos LTc a su vez se eliminan ellos mismos mediante lisis celular mediada por perforinas (fractricidio). En la LHH postrasplante se postula que el uso de medicamentos inmunosupresores induce un deterioro del fracticidio y la eliminación de las células presentadoras de antígenos (vías señaladas con “X”), lo cual perpetúa la respuesta inmune. Figura 1b: esquema de la citotoxicidad mediada por gránulos, en esta se observan las proteínas presentes en los gránulos citotóxicos de los LTc y la liberación de estos hacia las células diana. LTc: linfocito. C citotóxico. CD: célula presentadora de antígenos. Gz: granzimas. Perf: perforinas. Fuente: Modificado de Morimoto et al. y Voskoboinik et al. 4,30.
La activación de las NK y LTc favorece, además, la liberación de citoquinas proinflamatorias (interferón γ, factor de necrosis tumoral α, interleucinas IL-1b, IL-2, IL-6, IL-12, IL-18, entre otras) que estimulan a otras células, entre ellos los macrófagos, los cuales incrementan la activación de los LTc. Esta repuesta citotóxica está limitada mediante diferentes mecanismos, siendo el fratricidio (muerte entre LTc mediante lisis celular mediada por perforinas) uno de los mayores reguladores. Diferentes estudios han identificado como causantes de las LHH hereditarias (primarias) las mutaciones en genes relacionados con la síntesis o secreción de los gránulos citotóxicos de los LTc y las células NK. Por el contrario, en las LHH secundarias no es clara la causa que desencadena este proceso. Se postula que el uso de medicamentos inmunosupresores induce un deterioro de la actividad citotóxica de las células NK y los LTc para eliminar los procesos infecciosos o tumorales y un bloqueo del fratricidio 31; lo que lleva a una expansión no controlada del LTc con la consecuente perpetuación de la respuesta inmune y la secreción continua de citoquinas, activando macrófagos que a su vez producen más citoquinas para terminar, finalmente, en una tormenta de citoquinas con un grave síndrome de respuesta inflamatoria y falla orgánica multisistémica (Figura 1a) 17,30-37.
MANIFESTACIONES CLÍNICAS
Por lo general, el cuadro clínico se instaura de forma aguda ocurriendo en las primeras semanas luego del trasplante renal, cuando el paciente está intensamente inmunosuprimido, o también luego de recibir el tratamiento para el rechazo agudo 14. En algunos pacientes la LHH ha sido reportada años después del trasplante, asociada a la presencia de neoplasias, parasitosis, infección por parvovirus B19 e histoplasmosis 17. Los síntomas y signos más representativos son el marcado compromiso del estado general: fiebre alta, pérdida de peso, anorexia, linfadenopatías generalizadas y hepatoesplenomegalia por infiltración de macrófagos 15,21,38. Con menos frecuencia se encuentra rash cutáneo, púrpura, edemas, compromiso del sistema nervioso central que puede ser diverso, manifestándose como confusión, cefalea, meningitis, convulsiones y encefalopatía 1,4,29. En una serie de 17 pacientes con trasplante renal y diagnóstico de LHH, 7 de ellos presentaron síntomas neurológicos, siendo el estupor y las convulsiones los más frecuentes 15. También puede presentarse un compromiso del injerto renal con un cuadro histológico muy variado, como se describe a continuación.
Lesiones tubulointersticiales: son las más frecuentes y se caracterizan por el desarrollo de necrosis tubular aguda, con marcado infiltrado inflamatorio y la presencia de hemofagocitos. Entre el 30-50 % de los pacientes desarrollan una lesión renal aguda que se agrava con el uso de medicamentos nefrotóxicos y la falla multisistémica. Los estudios concluyen que la hipercitoquinemia, especialmente el incremento en los niveles del factor de necrosis tumoral α (FNT- α), produce apoptosis tubular a través de las vías de señalización ASK1 (apoptosis signal-regulating kinase 1) 2,39.
Lesiones glomerulares: entre los hallazgos histológicos reportados se encuentran la glomerulopatía variedad colapsante y la enfermedad de cambios mínimos. Adicionalmente, se observa una pérdida de la arquitectura de los podocitos sin acúmulos de complejos inmunes. También el FNT- α está aumentado, favoreciendo indirectamente los factores de transcripción y reorganización del citoesqueleto de actina 40,41.
Lesiones vasculares: el prototipo es la microangiopatía trombótica secundaria a una lesión endotelial por la tormenta de citoquinas 2,39.
ESTUDIOS DIAGNÓSTICOS
El diagnóstico de la LHH es un reto, ya que los hallazgos clínicos y de laboratorio pueden ser similares a los observados en otras enfermedades como infecciones, rechazo, cáncer o los efectos secundarios de los inmunosupresores u otros medicamentos. Todo esto obstaculiza el diagnóstico precoz de esta entidad 17. Con respecto a los exámenes de laboratorio, la hiperferritinemia es considerada uno de los hallazgos más útiles para el diagnóstico de LHH, especialmente cuando supera 10.000 ug/ml, con una sensibilidad y especificidad de 90 y 96 %, respectivamente 2,42,43. En el hemograma se puede encontrar un compromiso de dos o tres de las líneas celulares. Se presenta además hipertrigliceridemia, la cual es secundaria a la inhibición de la lipoproteinlipasa endotelial por el FNT- α. Dicha enzima que tiene como función la hidrólisis de triglicéridos para formar quilomicrones y VLDL.
Por lesión tisular puede haber un incremento de la deshidrogenasa láctica (LDH) y la creatinkinasa (CPK). Las pérdidas de líquidos por lesiones capilares inducen a hipoalbuminemia e hiponatremias de grado variable 1,4,5,29.
En laboratorios clínicos de referencia se pueden analizar los niveles del receptor soluble de la interleucina 2 (sIL2-R) o CD25, el cual es un marcador de la activación de linfocitos. Para su diagnóstico se precisan valores mayores a 2.400 UI/ml. La citometría de flujo se puede utilizar para evaluar la actividad de las NK, los niveles de perforinas en los LTc y el marcador de membrana de células citotóxicas CD107a, que hace parte de los gránulos que liberan Perf y Gz 1,29,44,45. En la biopsia de médula ósea se puede observar la presencia de eritrofagocitosis medular, hallazgo característico de la LHH 14; sin embargo, su ausencia no excluye el diagnóstico. En caso de una alta sospecha, se recomienda la toma de biopsias de médula ósea seriadas (Figura 2) 45. Adicionalmente, esta hemofagocitosis se puede observar en el tejido esplénico, hepático, renal, los ganglios linfáticos y la piel 14,44.
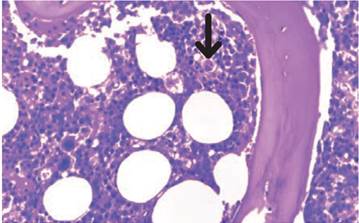
Figura 2 Hallazgos histológicos en la médula ósea. Biopsia de médula ósea (Giemsa, 40X), se observa la médula ósea hipocelular con la presencia de hemofagocito (flecha negra). Fuente: consentimiento, Nieto-Ríos et al. 8.
Ante la sospecha de compromiso neurológico se recomienda realizar, -a menos que la condición clínica la contraindique-, una punción lumbar para evaluar en el líquido cefalorraquídeo la presencia de pleocitosis (células mononucleares), proteinorraquia y, raramente, hemofagocitos 1. Otros de los hallazgos de los laboratorios reportados en pacientes con LHH postrasplante renal son las transaminasas elevadas, la hiperbilirrubinemia y alteración leve de las pruebas de coagulación 15.
La sociedad internacional del histiocito, en el 2004, actualizó los criterios clínicos y de laboratorio para el diagnóstico de LHH, el cual se hace con la confirmación molecular de las alteraciones genéticas o la presencia de 5 de los 8 criterios enlistados en la Tabla 4 4,46.
Tabla 4 Criterios diagnósticos para LHH (2004)
| A. Diagnóstico molecular consistente con LHH (PRF1, UNC13D, STXBP2, RAB27A, STX11, SH2D1A o XIAP) |
| B. Presencia de cinco de los ocho criterios clínicos siguientes: Fiebre Esplenomegalia Bicitopenia Hipertrigliceridemia (> 265 mg/dl) o hipofibrinogenemia (< 1,5 g/l) Hemofagocitosis Actividad células NK, baja o ausente Hiperferritinemia (> 500 μg / l) Aumento de los niveles de CD25 solubles (receptor IL-2) en suero (> 2,400 UI / ml) |
Posterior al diagnóstico, es fundamental encontrar la causa que desencadenó la LHH 17. La primera causa para considerar en la población trasplantada son las infecciones (virales, bacterianas, micobacterianas, micóticas, parasitarias). Para esto, se deben realizar los estudios microbiológicos respectivos que incluyen policultivos (sangre, médula ósea, tracto respiratorio, urinario, gastrointestinal), pruebas moleculares (cargas virales) y biopsias, entre otros. En la población colombiana con trasplante de riñón la causa más frecuente de LHH es la histoplasmosis diseminada 8-10. Otras posibles causas que deben ser evaluadas son la presencia de tumores malignos, en especial la enfermedad linfoproliferativa asociada al trasplante 24,25. Para dicha evaluación se deben realizar estudios imaginológicos corporales totales (TAC, resonancias, PET-CT), estudios moleculares del EBV, tanto en la sangre como en el tejido, estudios complementarios de médula ósea y biopsias de ganglios linfáticos u órganos comprometidos 24,25. De no encontrarse la causa, se deben considerar las enfermedades autoinmunes, la enfermedad del injerto contra huésped y, por último, las causas tóxicas o medicamentosas. En la población infantil se debe tener en cuenta desde el principio las causas genéticas 24.
TRATAMIENTO
La meta del tratamiento de la LHH es establecer un diagnóstico temprano antes de que se instaure la falla orgánica multisistémica. Los objetivos están encaminados a controlar la enfermedad de base (infección, neoplasia maligna, autoinmunidad, toxicidad, genética), atenuar a los macrófagos activados y limitar el estado de hiperinflamación 17. De no encontrarse prontamente la causa de la LHH y de no instaurarse un tratamiento oportuno y eficaz para esta causa encontrada, el resultado final será la falla multisistémica y la muerte del paciente 10.
El tratamiento coadyuvante de la LHH en los pacientes trasplantados de riñón se basa en la reducción de la dosis de inmunosupresores, vigilando la presencia de episodios de rechazo y el adecuado control de las infecciones con el manejo antimicrobiano apropiado. Se recomienda además, aunque sin mucha evidencia, el uso de esteroides a altas dosis con tiempos de duración no claramente establecidos 2,39. Existen escasos reportes del uso de la inmunoglobulina polivalente a dosis de 0,5 g/k una vez por semana, lo que podría ayudar en el manejo 1,47.
La quimioterapia con etopósido está reservada para casos muy esporádicos que son resistentes al manejo y para las formas genéticas 4,6,48. Estos pacientes podrían beneficiarse del trasplante de médula ósea, con posibilidad de supervivencia a los 3 años del 57 %, en los casos familiares 48. Algunos grupos han implementado terapias de rescate con plasmaféresis para la eliminación de citoquinas 4,17,49,50, biológicos inhibidores del FNT- α (etanercep, infliximab, adalimubab, golimubab, certolizumab) y alemtuzumab, anticuerpo monoclonal humanizado contra la glicoproteína de superficie celular CD52 con resultados variables 51.
Infortunadamente, el tratamiento de la LHH aún no está definido y los estudios donde se han evaluado pacientes trasplantados renales con LHH son muy heterogéneos, con un bajo número de pacientes y diferentes combinaciones de medicamentos; por lo que la decisión terapéutica, en muchas ocasiones, se basa en la experiencia más que en los datos disponibles. Se sugiere en estos pacientes la búsqueda de las causas que llevaron a esta enfermedad, sumado a la terapia coadyuvante, todo esto encaminado a reducir la mortalidad de la LHH.
CONCLUSIÓN
La LHH es una complicación rara que puede presentarse posteriormente al trasplante renal, esta se relaciona generalmente, con causas secundarias como los medicamentos inmunosupresores, episodios de rechazo, las infecciones, neoplasias, tóxicos y enfermedades autoinmunes, entre otros. El curso clínico generalmente es fulminante y se produce alta morbimortalidad; por lo cual esta entidad se debe sospechar tempranamente, cuando los pacientes presentan fiebre, organomegalias y/o citopenias. En los estudios complementarios usualmente se encuentra hiperferritinemia, hipertrigliceridemia, hipofibrinogenemia, hemofagocitosis, elevación del CD25 soluble y una alteración en la actividad de las células NK. En la actualidad no existe un tratamiento definitivo, por este motivo hacemos énfasis en la necesidad de un diagnóstico precoz y el control oportuno de la causa desencadenante.

