











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La primera asociación clínica entre el carcinoma medular de tiroides (CMT) y el feocromocitoma fue descrita en 1961 por Sipple 1. El término neoplasia endocrina múltiple tipo 2 (MEN 2, por sus siglas en inglés) fue propuesto por Steiner en 1968 2, y solo cinco años después se informó sobre dos pacientes con neuromas de la mucosa oral, CMT y feocromocitoma 3. La descripción de una rara entidad que agrupa CMT hereditario con un fenotipo de neuroma mucoso fue finalmente nombrada MEN 2B por Chong et al. en 1975 4.
MEN 2B, también conocido como síndrome de Wagenmann-Froboese, es una enfermedad tumoral hereditaria autosómica dominante, causada por mutaciones de la línea germinal en el protooncogén RET. La mutación más frecuente (>95 % de los casos) es una sustitución de metionina por treonina en el codón 918 (M918T), y representa entre el 5 % y el 20 % de las formas de MEN 2. Se caracteriza por la asociación con CMT en el 100 % de los pacientes, feocromocitomas en el 50 % (de los cuales aproximadamente la mitad son múltiples y bilaterales), hábito marfanoide hasta en el 97 % de los afectados y ganglioneuromatosis intestinal y neuromas mucosos en más del 90 % 5.
Los rasgos fenotípicos de MEN 2B en comparación con MEN 2A incluyen una presentación más agresiva y temprana del CMT en la primera y segunda década de la vida, características extraendocrinas prominentes y la ausencia de hiperparatiroidismo primario. Debido a su muy baja prevalencia, de 0,9 a 1,65 por millón de habitantes, e incidencia de 1,4 a 2,6 por millón de nacidos vivos por año 6,7, la historia natural de este síndrome sigue estando pobremente descrita en la literatura 8.
Bases genéticas en men 2B
Desde el descubrimiento del gen RET en 1985 9 se ha logrado un progreso considerable en la comprensión de su fisiología y papel en las enfermedades, particularmente en diversos subtipos de cáncer de tiroides.
El protooncogén RET se encuentra en el cromosoma 10q11.2 y tiene 21 exones que abarcan 55kb; su dominio extracelular consta de una región rica en cisteína cerca de la membrana celular y un dominio similar a cadherina más distal. Más allá de una porción transmembrana hay una intracelular con dos dominios tirosina quinasas 10.
Mutaciones de sustitución en uno de los cinco residuos de cisteína en los exones 10 y 11 en el dominio extracelular conducen a la neoplasia endocrina múltiple tipo 2A (MEN 2A) y al carcinoma medular de tiroides familiar (CMTF). Las mutaciones en los codones 768 y 883 en los exones 13 y 14 de la región de tirosina quinasa 1 intracelular están asociadas con CMTF aislado. La mutación de metionina a treonina en el codón 918 en el exón 16 de la región de tirosina quinasa 2 intracelular da como resultado MEN 2B 11 (Figura 1).
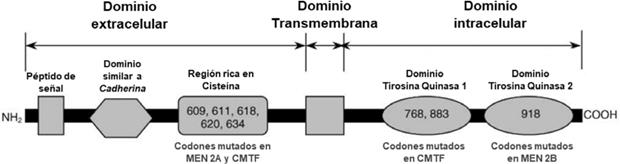
Fuente: elaboración propia
Figura 1. Diagrama esquemático del protooncogén RET. Mutaciones que dan como resultado síndromes congénitos asociado a CMT.
La unión del ligando RET causa dimerización y activación del receptor. El gen RET es importante en cascadas de señalización para el crecimiento celular y la diferenciación de tejidos derivados de las células de la cresta neural, como las células C parafoliculares de la tiroides, médula suprarrenal, glándula paratiroidea y el plexo nervioso autónomo entérico 11. Las variantes del gen RET están presentes en el 98 % de los pacientes con MEN 2. MEN 2B se debe a mutaciones de novo de la línea germinal activante autosómica dominante de este protooncogén RET 12.
Se han detectado correlaciones significativas de genotipo-fenotipo. La mutación M918T es responsable de más del 95 % de los casos de MEN 2B 13,14, ocurre en el dominio tirosina intracelular del gen RET y conduce a hiperplasia de células C de la tiroides. Existe una asociación entre la presencia de la variante y un peor pronóstico en CMT 15,16. La mayor agresividad de M918T puede deberse, en parte, a la ubicación del aminoácido 918 dentro del núcleo catalítico de la quinasa RET, que conduce a un aumento en la unión de ATP y la actividad enzimática de la quinasa y la capacidad para ser activado sin la dimerización del receptor. El receptor mutado M918T puede activarse aún más por su ligando endógeno, incluso si ya está hiperactivado espontáneamente, y esto podría explicar que se dé una presentación clínica más agresiva, o al menos una edad muy temprana del diagnóstico de CMT 17,18.
También se han informado otras mutaciones raras que involucran al codón 883 (A883F) o doble heterocigotos en menos del 5 % de los casos, con sustitución de alanina por fenilalanina. Otras, como las presentes en el codón 804 y codón 883, sugieren un fenotipo del CMT menos agresivo en comparación con los pacientes con la mutación del codón M918T 19.
Un grupo raro de pacientes tiene MEN 2B atípico que se desarrolla alrededor de los 20 a 30 años y presentan compromiso de la línea germinal de RET dobles que aparecen en tándem en el mismo alelo, e involucran el codón V804M de RET y el codón Y806C, S904C, E805K o Q781R de RET. La evaluación de las mutaciones en tándem mediante análisis in vitro e in silico proporciona información sobre su capacidad de transformación (puntuaciones de predicción). Se han identificado variantes similares de la línea germinal de RET doble en familias con CMTF de MEN 2A 20.
Aunque existe una distribución por sexos equitativa en la presentación hereditaria, Carlson et al. han demostrado que el 100 % de los casos esporádicos de MEN 2B se formaron a partir de compromiso en el cromosoma de origen paterno, lo que sugiere que un alelo RET puede ser más susceptible a mutación cuando se hereda del padre y, también, que estas están asociadas con la edad paterna avanzada, lo que sugiere una susceptibilidad diferencial de RET a la mutación en el ADN derivado de progenitores 11.
El manejo jerárquico de genes del CMT relacionado con MEN 2B puede facilitar en gran medida una mejoría del pronóstico. Según el sitio de mutación de RET, la American Thyroid Association (ATA) dividió el riesgo en tres categorías según su agresividad: muy alto riesgo, alto riesgo y riesgo moderado, basadas en el desarrollo de CMT a edades tempranas y la asociación con enfermedad metastásica (21,26) (Tabla 1).
Tabla 1 Relación de mutaciones de RET con el riesgo de agresividad del CMT e incidencia de feocromocitoma en MEN 2B según ATA
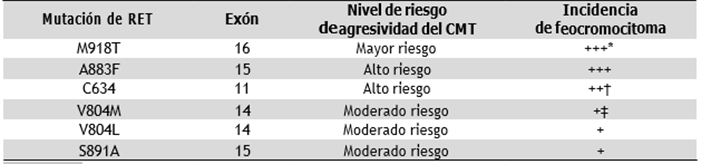
*+++: ~50 %
†++: ~20-30 %
‡+: ~10 %
Fuente: elaboración propia
Esencialmente, existe una penetración completa del defecto genético, aunque con expresividad variable, ya que no todos los pacientes manifiestan todas las características del síndrome 22. Como RET es un protooncogén, una sola mutación de la línea germinal debería ser suficiente para producir CMT. Sin embargo, la variabilidad fenotípica en MEN 2B podría explicarse por la aparición de mutaciones somáticas asociadas que podrían acelerar el proceso oncogénico y modificar la agresividad de CMT 19,23. Recientemente un perfil completo de expresión génica de muestras de CMT de MEN 2A y MEN 2B identificó tres genes desregulados de manera diferente, lo que podría explicar los diferentes fenotipos: NNAT (gen supresor de tumores), CD14B (control del ciclo celular) y NTRK3 (receptor de tirosina quinasa) 24.
Finalmente, la mutación RET subyacente también determina las características extratiroideas de MEN 2, probablemente impartiendo una susceptibilidad diferente de las células suprarrenales o paratiroideas a un receptor mutante dado. Esto podría explicar la falta de hiperparatiroidismo primario en MEN 2B o, por ejemplo, la diferencia en la prevalencia de feocromocitoma en pacientes M918T en comparación con aquellos que albergan mutaciones RET de la línea germinal del exón 10, que también se correlaciona con la presentación de enfermedad de Hirschsprung 19.
El conocimiento de estas mutaciones ha permitido identificar a los individuos susceptibles de desarrollar la enfermedad, así como confirmar el diagnóstico, correlacionar el genotipo con el fenotipo o su expresión clínica, estratificar el riesgo de fallecer o la agresividad de la enfermedad, y establecer el tratamiento y el seguimiento más apropiados. Por lo anterior, la asesoría genética y la evaluación del protooncogén RET es rutinaria en el enfoque del MEN 2B 25.
La guía para el tratamiento del CMT de la American Thyroid Association (ATA) recomienda que todos los pacientes con el fenotipo MEN 2B deben someterse a pruebas genéticas para la mutación del codón RET M918T (exón 16) y, si esta es negativa, también para compromiso del codón RET A883F (exón 15). Si no se identifican en estos dos exones, se debe secuenciar toda la región codificante de RET 26. Lo anterior, considerando que la inhibición de la quinasa RET es una diana terapéutica atractiva en pacientes con mutaciones genéticas asociadas a MEN 2B mediante el uso de inhibidores quinasas 20.
Componentes y manifestaciones de la enfermedad
1. Carcinoma medular de tiroides
El CMT se origina en las células C derivadas de la cresta neural de la glándula tiroides, las cuales tienen capacidad secretora hormonal. La secreción de calcitonina es la más característica, aunque también pueden secretar prostaglandinas, ACTH, antígeno carcinoembrionario (ACE), somatostatina, neurotensina, sustancia P e histamina. La investigación sobre CMT durante los últimos 55 años ha permitido comprender bien los defectos genéticos y las vías moleculares alteradas asociadas con su desarrollo, y con ello identificar y tratar tempranamente a pacientes con CMT esporádicos o hereditarios 27,28.
En pacientes con MEN 2B, el CMT se desarrolla durante los primeros años de vida en el 100 % de los casos, y sigue siendo la principal causa de muerte. Por lo general, es la primera enfermedad que se diagnostica, en promedio en la segunda década de la vida, unos 10 años antes de lo observado en individuos con MEN 2A 29. En comparación con CMT esporádicos, el que se encuentra asociado a MEN 2B generalmente es multifocal, bilateral y acompañado de hiperplasia de células C. Los pacientes con MEN 2B tienen un peor pronóstico, metástasis más precoces y una supervivencia a 10 años del 75,5 % en comparación con el 97,4 % en MEN 2A 30. El compromiso metastásico está presente en un 45 % de los casos al momento del diagnóstico, y en un 50 % se documenta durante la cirugía. Inicialmente son ganglionares cervicales y mediastinales, aunque por vía hematógena pueden generar diseminación a distancia al hígado, pulmón y huesos 31.
Por lo general, aquellos con MEN 2B que no se diagnostican con base en el fenotipo típico debutan con una masa en cuello que se correlaciona imagenológicamente con una neoplasia dependiente de tiroides asociada a una concentración plasmática elevada de calcitonina y ACE. Todos los pacientes con CMT sospechado o confirmado deben someterse a una ecografía de cuello. Si hay evidencia de compromiso ganglionar adyacente y el paciente tiene elevación preoperatoria marcada de calcitonina (>500 pg/mL) se debe realizar una tomografía contrastada de tres fases de cuello y tórax, resonancia magnética nuclear contrastada de hígado y gammagrafía ósea de esqueleto axial 32).
La sensibilidad de estas técnicas oscila entre el 25 % y el 50 %, por lo cual los estudios radiológicos recientes se han centrado en probar diferentes radiotrazadores y modalidades que podrían proporcionar mayor sensibilidad y eficiencia para pacientes con niveles elevados de calcitonina y ACE. Entre ellos, la tomografía por emisión de positrones (PET) con fludesoxiglucosa F 18 (18F-FDG) puede detectar la recurrencia tumoral con sensibilidades informadas entre el 15 % y el 78 % y basadas en lesiones que oscilan entre el 28 % y el 96 %. La obtención de imágenes por PET con radiotrazador 18F-dihidroxifenilalanina (DOPA) ha aumentado la sensibilidad en comparación con 18F-FDG. Las células tumorales neuroendocrinas como el CMT pueden absorber y descarboxilar precursores de amina, como DOPA, lo que permite la visualización por PET con 18F-DOPA. Múltiples estudios comparativos informan una mejor sensibilidad para la 18F-DOPA, entre el 63 % y el 87 % 28.
Dada la secreción hormonal del CMT, los pacientes podrían presentar diarrea como manifestación clínica hasta en el 30 % de los casos 21. El diagnóstico histológico está dado por la presencia de nidos de células C con multinucleación, amiloide y calcificaciones, así como trabéculas, patrones de crecimiento tumoral de células alveolares y fusiformes que parecen extenderse más allá de la membrana basal e infiltrar y destruir los folículos tiroideos. Se ha descrito la hiperplasia de células C como un precursor del CMT presente en etapas aún más tempranas de la vida en pacientes con MEN 2B. La inmunohistoquímica para la expresión de calcitonina y ACE se puede realizar como un complemento de diagnóstico patológico 28,33.
En pacientes con enfermedad metastásica a distancia avanzada estructuralmente evidente (ya sea en la presentación inicial o después de un período prolongado de latencia), los tiempos de duplicación de los marcadores tumorales para niveles de calcitonina o ACE de hasta 12 meses identifican pacientes con altas tasas de mortalidad dentro de los siguientes 5 años 34.
La tiroidectomía total temprana es el tratamiento de primera línea del CMT asociada a MEN 2B, ya que puede modificar el curso clínico y el pronóstico a largo plazo de la enfermedad. La terapia quirúrgica en el CMT hereditario es el mismo que el del CMT esporádico 26. En todos los pacientes con un diagnóstico preoperatorio de cáncer de tiroides se debe realizar una búsqueda exhaustiva de linfadenopatías cervicales previo a la intervención quirúrgica 34. Además, después de la primera cirugía se puede presentar enfermedad residual o recurrencia hasta en el 83 % de los casos 35.
La tiroidectomía profiláctica (en portadores presintomáticos) antes de la aparición de CMT fue recomendada por primera vez en pacientes con MEN 2, hace más de 20 años, por Wells et al. 37; Skinner et al. 38 confirmaron la viabilidad del enfoque 10 años después. El objetivo oncológico de la cirugía temprana no es tanto prevenir que ocurra la malignidad en primer lugar, sino principalmente extirpar la tiroides antes de que ocurran las metástasis; los datos recientes sugieren que esto se puede lograr en la mayoría de los pacientes con MEN 2B tratados antes de los 4 años 14. Sin embargo, en la práctica muy pocos niños con MEN 2B son sometidos a una verdadera tiroidectomía profiláctica, ya que esta es realizada en una enfermedad clínica manifiesta 35. Por lo anterior, la educación adecuada de pediatras, oftalmólogos, dentistas, gastroenterólogos, ortopedistas y médicos generales respecto a la identificación de MEN 2B es crucial, ya que representan la primera línea para lograr un diagnóstico precoz 36.
En el estudio más grande publicado sobre MEN 2B se incluyó un total de 345 pacientes, de los cuales el 21 % (n = 71) falleció a una mediana de edad de 25 años (rango <1-59). El 98 % (n = 338) se sometió a una tiroidectomía total realizada antes del primer año de vida (n = 20), lo cual condujo en el 83 % de los casos a una remisión a largo plazo (es decir, nivel de calcitonina indetectable). Las curvas de supervivencia específicas del CMT no mostraron ninguna diferencia significativa entre aquellos que se sometieron a tiroidectomía antes o después del primer año, pero sí una diferencia estadísticamente significativa en el estado de remisión en este grupo. En los otros 318 pacientes que se sometieron al procedimiento después del primer año la remisión se obtuvo en el 15 % (47 de 318 individuos), lo que lleva a concluir que la tiroidectomía realizada antes de dicha edad se asocia con una alta probabilidad de curación 36.
Las correlaciones genotipo-fenotipo han facilitado el desarrollo de pautas para las intervenciones. Los enfoques actuales para decidir la edad apropiada a la que se debe realizar la cirugía incorporan datos de la ecografía y las mediciones de calcitonina, además de los resultados de la genotipificación 39.
La probabilidad de curación quirúrgica es indudablemente menor en MEN 2B que en MEN 2A, probablemente por una edad más prematura de aparición de CMT y un diagnóstico clínico tardío 40. Para evitar el riesgo de desarrollar un CMT incurable, las pautas actuales de la American Thyroid Association recomiendan que en niños portadores de la mutación M918T se realice la tiroidectomía temprana antes de que cumplan un año. Cuando el diagnóstico se realiza después de esta edad, se debe realizar, además de la tiroidectomía total, disección cervical central profiláctica, siempre y cuando se asegure la función paratiroidea y la cirugía se realice en un centro experimentado 26. Sin embargo, a excepción de los casos familiares, esto es difícil de lograr en la práctica actual, ya que la gran mayoría de los pacientes con MEN 2B portan mutaciones de novo, lo que lleva a un diagnóstico tardío y dificulta la intervención temprana 35.
Contrario a lo que ocurre con el carcinoma diferenciado de tiroides (papilar y folicular), las células del CMT no captan el yodo y, por consiguiente, tal terapia no tiene efectividad en esta enfermedad. Una vez está avanzada y se han agotado las alternativas de manejo (como radioterapia, radiocirugía, ablación con radiofrecuencia o embolización de las metástasis), las opciones disponibles estarán reservadas para los inhibidores de la tirosina quinasa 32.
Los inhibidores de la tirosina quinasa (ITK) se constituyen como un gran avance para el tratamiento sistémico de pacientes con cáncer de tiroides avanzado, en particular CMT. La inhibición de un solo receptor puede inducir la activación compensadora de otras, por lo que su bloqueo simultáneo de diferentes tirosinas quinasa activadas puede ser la mejor manera de abordar el CMT (27). Los ITK anti-RET que alcanzaron el desarrollo clínico han demostrado una actividad considerable contra el receptor del factor de crecimiento endotelial vascular tipo 2 (VEGFR2) y otros miembros de la familia VEGFR. Por lo anterior, los ensayos en curso están enfocados en moléculas de nueva generación más potentes y selectivas para inhibición de RET 10.
A la fecha, las agencias regulatorias han aprobado el uso de inhibidores multiquinasa, como el vandetanib y cabozantinib, y los inhibidores de RET selpercatinib y pralsetinib. Vandetanib pertenece a los inhibidores de la tirosina quinasa tipo I, definidos como inhibidores que se unen al ATP con el segmento de activación en la conformación activa. En estudios celulares, éste fármaco inhibió la actividad de los productos de fusión RET, así como mutaciones puntuales de RET 10. Adicionalmente, se observó que inhibe selectivamente al VEGFR-2 y 3 y EGFR 21, con lo cual inhibe la proliferación, la supervivencia y la angiogénesis de las células tumorales sin provocar efectos citotóxicos directos sobre las células endoteliales 27.
Wells et al. informaron en 2012 los resultados de un estudio de fase III aleatorizado doble ciego, controlado con placebo sobre vandetanib, en 331 pacientes con CMT metastásico (estudio ZETA). La mediana de supervivencia libre de progresión fue significativamente superior en el grupo de vandetanib frente a placebo (30,5 [predicho] frente a 19,3 meses, respectivamente, [p < 0,001]).
Específicamente en el subgrupo de casos con CMT no hereditario y una mutación somática M918T (n = 101), se observó una tasa de respuesta objetiva del 54,5 %. El 55 % de los pacientes en el ensayo ZETA presentó eventos adversos, entre los cuales la diarrea fue el más común (56 %), seguida de anomalías cutáneas (erupción cutánea, acné y foliculitis). Otros efectos secundarios frecuentes fueron dolor abdominal, fatiga, cefalea, hipertensión, disminución del apetito y náuseas, que ocurrieron en el 20 % al 30 % de los pacientes. El efecto secundario más preocupante fue la prolongación del intervalo QT (presente en el 14 %), que puede predisponer a los pacientes a arritmias como torsade de pointes y muerte súbita 41.
Fox et al. también informaron en el 2013 del primer ensayo clínico de fase I/II de vandetanib en 16 niños y adolescentes con CMT metastásico, 15 de los cuales tenían MEN 2B y mutación M918T. Los 15 pacientes tuvieron una disminución en el tamaño del tumor y 7 de ellos una respuesta parcial confirmada 42. Los estudios preclínicos sugieren que el CMT asociado con la mutación RET V804M es resistente al vandetanib. Aunque estos casos presentan un curso clínico agresivo, son raros y la experiencia con la terapia sistémica en ellos es limitada 34.
Por otro lado, cabozantinib es un inhibidor de la tirosina quinasa tipo II, definido como inhibidores que se unen a la conformación catalíticamente inactiva de la quinasa 10. Es un inhibidor multicinasa de VEGFR1, VEGFR2, RET y c-MET 21. En un estudio de fase III controlado con placebo doble ciego aleatorizado por Elisei et al. en 2013 (estudio EXAM) con 330 pacientes, cabozantinib mostró una mejoría significativa de la mediana de supervivencia libre de progresión (11,2 vs. 4,0 meses en los grupos de intervención y placebo, respectivamente).
El efecto secundario más común informado en el EXAM fue diarrea (63 %), seguida de elevación de las concentraciones séricas de TSH, hipocalcemia, estomatitis, síndrome mano-pie, pérdida de peso, náuseas, hiporexia y fatiga (40-50 %) 43. Sherman et al. informaron posteriormente, en un análisis de subgrupos del ensayo de fase III, que los pacientes con una mutación M918T tuvieron un mayor beneficio de supervivencia libre de progresión cuando se manejaron con cabozantinib 44.
La selección de la terapia sistémica de primera línea (cabozantinib frente a vandetanib) para el CMT avanzado en pacientes con MEN 2B debe ser individualizada. El estado funcional del paciente, la edad, las comorbilidades, las condiciones sociales, la progresión de la enfermedad, el examen físico, la disponibilidad de cada medicamento, y especialmente la seguridad, son aspectos importantes para considerar a la hora de decidir cuál administrar. La experiencia clínica sugiere que vandetanib es más fácil de tolerar que cabozantinib; sin embargo, este último, por su potencial antiangiogénico, es más potente. Por lo tanto, cabozantinib puede ser el tratamiento más eficaz en pacientes con CMT que progresa rápidamente 34.
En estudios pequeños se han investigado otros inhibidores de tirosina quinasa como axitinib, sorafenib, sunitinib, pazopanib y motesanib con respuestas parciales o estabilización de la enfermedad, aunque no se ha avanzado en su desarrollo clínico 21.
El bloqueo simultáneo de la quinasa RET y VEGFR2 podría proporcionar una ventaja en el control del crecimiento tumoral, aunque tal actividad dual incrementa la toxicidad de los ITK y, por lo tanto, obstaculiza la posibilidad de lograr dosis seguras que sean suficientes para inhibir la RET quinasa. En ese sentido, los esfuerzos de investigación han llevado a la identificación de nuevos ITK con actividad inhibitoria variable de VEGFR.
Pralsetinib es un inhibidor potente y altamente selectivo de RET, con una potencia 10 veces mayor y con una tasa de respuesta objetiva del 58 % en pacientes con fusión RET positiva. Selpercatinib es una molécula pequeña que inhibe el RET de manera competitiva con el ATP. Es altamente selectivo y potente, y puede inhibir mutaciones que predicen resistencia a otros inhibidores multiquinasa, con una tasa de respuesta objetiva de hasta el 68 % en pacientes con fusión de RET. Estos dos inhibidores de RET han sido aprobados para el uso en carcinoma medular de tiroides por la FDA y la EMA; no obstante, no están disponibles en Colombia a la fecha de esta publicación.
Otro potente ITK de RET es BOS172738, que tiene una alta selectividad sobre VEGFR2 (>300 veces) y ha demostrado potencia contra varias mutaciones de RET. Finalmente, TPX-0046 es otro nuevo ITK que ahorra VEGFR2 con actividad inhibidora dual de la quinasa RET-SRC a concentraciones nanomolares bajas que demostraron tener eficacia antitumoral. Estas moléculas se encuentran actualmente en ensayos clínicos 10,32.
2. Feocromocitoma
El feocromocitoma es la segunda neoplasia más frecuente en pacientes con MEN 2B con una penetrancia del 40 % al 50 %, y se presenta como la primera manifestación en la minoría de los casos (<1 %). Puede aparecer de manera sincrónica con el CMT en el 35 %. La gran mayoría (>95 %) son benignos: sólo el 0 % al 4 % hacen metástasis 35. Se presentan de forma más tardía en el MEN 2B, entre la tercera y cuarta década de la vida, con una edad media de presentación de 28 años. Algunos estudios han mostrado su aparición en los diez años posteriores a la hiperplasia de células C o al CMT; generalmente son bilaterales y multicéntricos 45. La presentación de paragangliomas es extremadamente rara.
En el estudio más grande publicado de MEN 2B hasta el momento se incluyó un total de 345 pacientes, de los cuales se diagnosticó feocromocitoma bilateral en el 50 % (n = 156), con una media de edad al diagnóstico de 28 años. Se realizó cirugía de conservación suprarrenal en 31 pacientes, de los cuales el 10 % tuvo recurrencia a largo plazo, mientras que la función suprarrenal normal se obtuvo en un 62 % de los casos 36. En el estudio de Thosani et al., de 85 pacientes con MEN 2 y feocromocitoma, 70 tenían MEN 2A y 15 tenían MEN 2B. La mediana de edad al momento del diagnóstico de feocromocitoma fue 32 años. La manifestación inicial sincrónica de CMT y feocromocitoma se presentó en el 6 %. El 72 % tenían presentación bilateral y la mayoría de los tumores eran sincrónicos (82 %) 46.
Los signos y síntomas de feocromocitoma son iguales a los de pacientes sin MEN 2B. Sin embargo, entre el 30 % y el 50 % son asintomáticos y diagnosticados durante un programa de tamizaje. El diagnóstico es confirmado por la demostración de concentraciones elevadas de metanefrinas fraccionadas en plasma o en orina de 24 horas; típicamente secretan cantidades desproporcionadas de epinefrina 35.
La guía de la ATA para el tratamiento de pacientes con CMT recomienda que todo paciente con MEN 2B y un diagnóstico histológico de CMT, independientemente de la edad y los síntomas que presenten, debe tener un feocromocitoma excluido antes de cualquier procedimiento intervencionista 26.
Adicionalmente, para los pacientes con MEN 2B cuyos resultados de detección iniciales son negativos para feocromocitoma, la guía de manejo de la ATA recomienda que los estudios deben comenzar a los 11 años para los niños de las categorías ATA-HST (mayor riesgo: MEN 2B y la mutación del codón RET M918T) y ATA-H (alto riesgo: incluye pacientes con mutación del codón C634 de RET y la mutación del codón RET A883F) y a los 16 años en los niños de la categoría ATA-MOD (moderado riesgo: incluye pacientes con mutaciones del codón RET que no sean M918T, C634 y A883F) 26 (ver Tabla 1).
El cribado consiste en medir metanefrinas y normetanefrinas libres en plasma o metanefrinas y normetanefrinas urinarias de 24 horas. Las imágenes suprarrenales con tomografía o resonancia magnética nuclear están indicadas en pacientes con resultados bioquímicos positivos 26.
El manejo quirúrgico es la terapia de elección. Si se realiza un diagnóstico sincrónico con el CMT, deberá resecarse primero el feocromocitoma. El abordaje abierto o laparoscópico es apropiado, con desenlaces similares. Existe controversia acerca de la necesidad de adrenalectomía bilateral, en lo cual influye la posibilidad de malignización, desarrollo tumoral en la glándula contralateral e insuficiencia suprarrenal postoperatoria 47,48, por lo que no se recomienda de forma rutinaria en pacientes con enfermedad unilateral.
La adrenalectomía subtotal, en la cual se preserva entre el 10 % y el 30 % de tejido residual glandular, parece ofrecer una capacidad de estrés suprarrenal suficiente para obviar la necesidad de suplementos de corticosteroides en más del 80 %, y debe ser considerada en todos los pacientes con CMT antes de la cirugía (independientemente de los signos y síntomas) 49. Luego de la cirugía, los pacientes requieren seguimiento de por vida, debido al riesgo de recurrencia de feocromocitoma, el cual es de aproximadamente del 20 % dentro de los 20 años posteriores a la adrenalectomía subtotal 50.
3. Características extraendocrinas
Los pacientes con MEN 2B presentan un fenotipo clínico dado por las características extraendocrinas descritas (que pueden ser incompletas para cada caso determinado). Todos los pacientes con MEN 2B tendrán una o más de estas manifestaciones no endocrinas 29, por lo cual la asociación de varios de estos signos clínicos debe hacer sospechar el síndrome clínico en el prestador de salud, y con ello dar lugar a la tamización inicial para CMT.
En el estudio sobre MEN 2B más grande publicado a la fecha, todos los pacientes con datos disponibles (n = 287) tenían al menos una característica extraendocrina. El 56 % presentaba hábito marfanoide, neuromas de la mucosa y signos gastrointestinales 36. Hasta el momento de esta publicación, en Colombia solo se ha descrito en la literatura un reporte de caso de una paciente femenina de 8 años con MEN 2B asociado a una mutación en el codón 918 del exón 16 del protooncogén RET, asociado a CMT y neurinomas en lengua y cavidad oral 51. Al momento, Román-Gonzales et al. se encuentran describiendo la primera serie de pacientes con MEN 2B en Colombia.
3.1 Hábito corporal marfanoide y otras características esqueléticas
Se conoce con este nombre a las características físicas de los pacientes con MEN 2B que simulan un síndrome de Marfán. El fenotipo esquelético suele incluir una expresión variable de un hábito corporal marfanoide, caracterizado por una estatura alta, extremidades largas, una cara delgada y alargada y aracnodactilia de los dedos de manos y pies. También se pueden asociar otras anomalías esqueléticas como lordosis, cifosis, escoliosis, hipermovilidad articular, pie cavo, pectus excavatum (relacionado con el crecimiento excesivo de las costillas), paladar arqueado alto y deslizamiento epifisiario femoral 35.
3.2 Neuromas mucosos
Es el rasgo más característico de este síndrome, por lo que en la literatura también se le conoce como síndrome de neuromas mucosos 52. Casi todos los pacientes con MEN 2B desarrollan neuromas mucosos múltiples, los cuales se ubican en la región centrofacial, principalmente en el tercio anterior de la lengua, mucosa labial, cavidad oral, nasal, conjuntival, palpebral y, además, en todo el tubo digestivo. A menudo son el primer rasgo clínico en aparecer y pueden ser evidentes desde el nacimiento; ocurren en la mayoría de los casos dentro de la primera década de vida, aunque su aparición puede retrasarse hasta una edad avanzada (53 a 55 años).
Los neuromas de las mucosas se describen como múltiples pápulas pequeñas y blandas en la cavidad oral o alrededor de ella, que incluyen la punta de la lengua y los labios, las mucosas nasal y laríngea y las conjuntivas. Las lesiones aparecen como nódulos sésiles indoloros de 2 a 7 mm de color amarillo a blanco. Cuando hay un número suficiente, dan una apariencia de grasa 35. Histopatológicamente, los neuromas consisten en una proliferación encapsulada de nervio y fibras de colágeno y células paraneurales 56. Son neuromas verdaderos, por lo cual deben diferenciarse de los neurofibromas, asociados a la neurofibromatosis (Figura 2).
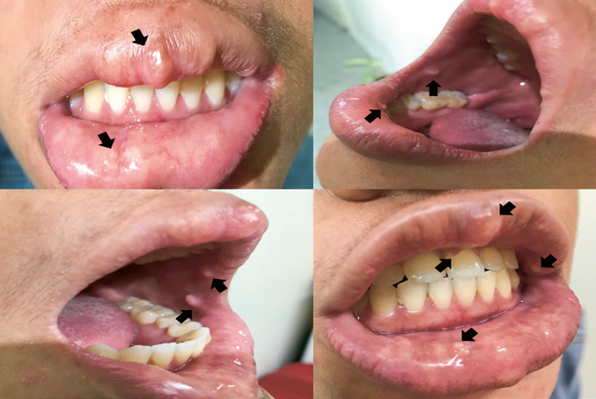
3.3 Signos oftalmológicos
La incidencia de anormalidades oculares es difícil de estimar, puesto que habitualmente no se describen. En la serie reportada por Brauckhoff et al. 13 estas manifestaciones estaban presentes en todos los pacientes con información clínica detallada; la alacrimia (llanto sin lágrimas) es uno de los más comunes. Otras características son la prominencia de nervios corneales, el aumento del volumen y eversión palpebral, ptosis leve, desplazamiento del conducto lagrimal y de pestañas, neuromas subconjuntivales, dilatación pupilar, aumento de la presión intraocular, xeroftalmia y xerostomía 57,58.
3.4 Signos gastrointestinales
Hace más de 40 años, Carney et al. se centraron por primera vez en los signos gastrointestinales asociados con MEN 2B 59. Especialmente la constipación, la intolerancia alimentaria y la seudoobstrucción intestinal suelen representar las primeras manifestaciones inespecíficas de MEN 2B, que se pueden observar desde la infancia y están presentes hasta en el 90 % de los pacientes. Por lo anterior, se debe considerar el síndrome de MEN 2B en el diagnóstico diferencial de niños con constipación crónica o seudoobstrucción intestinal 60.
Los neuromas pueden encontrarse en todo el tracto gastrointestinal en combinación con otras alteraciones del sistema ganglionar autónomo en la pared intestinal, como la existencia de un desorden neuropático difuso de la motilidad intestinal 61. Los pacientes afectados cursan con proliferación del plexo mientérico, causada por hiperplasia del tejido nervioso, incluyendo células ganglionares con sus células nerviosas y de Schwann. Esta proliferación se asocia a fibrosis, lo que permite la fácil distensión abdominal 54,55.
Los problemas gastrointestinales se deben a una ganglioneuromatosis intestinal difusa que se presenta en el 30 % de los casos de MEN 2B que, si bien es más común que se localice en el intestino delgado y el grueso, puede presentarse en otros sitios del tubo digestivo como el hígado, la vesícula biliar y el páncreas. A ello se asocia una motilidad colónica alterada que conduce a una seudoobstrucción intestinal y al desarrollo final de megacolon.
La ganglioneuromatosis en el tracto gastrointestinal da una sintomatología clínica de dolor abdominal tipo cólico, obstrucción intestinal, cambios en el hábito intestinal con constipación o diarrea. Además, se describe megacolon (31 % a 66 %), diverticulitis secundaria (6 % a 14 %) y disfagia en pequeño porcentaje. El adulto puede llegar a la cirugía de urgencia, siendo la causa más común la enfermedad diverticular complicada con perforación, y en niños la obstrucción intestinal 54. Es de destacar que el aumento de la secreción de catecolaminas (como se observaría en el feocromocitoma asociado a MEN 2B) puede empeorar la constipación y rara vez conduce a megacolon tóxico 60.
Los pacientes con MEN 2B también pueden presentarse con sintomatología gastrointestinal superior y manifestaciones esofágicas. Se ha descrito que hasta el 39 % de los casos presentan disfagia, y hasta el 14 % episodios eméticos. El tratamiento médico indicado se centra en ajustes en la dieta, laxantes y suplementos de fibra, lo cual puede ser difícil. En última instancia algunos pacientes (aproximadamente un tercio) pueden requerir hospitalización o cirugía 62,63.
Las manifestaciones intestinales también pueden ser predictivas de la agresividad del CMT, como sugieren algunos autores que muestran que un peor pronóstico del CMT se asoció con signos gastrointestinales más graves 35,64.
3.5 Otros signos
Se han informado otras asociaciones en MEN 2B, entre las que se incluyen facies toscas, mala posición dental, pies anormales con un primer dedo largo y un espacio aumentado entre el primer y el segundo dedo, malformaciones renales y bajo peso 65. Se ha descrito neuropatía periférica, hipotonía muscular, hiperhidrosis facial, atrofia testicular y pubertad retrasada asociada a MEN 2B 66.
Conclusiones
El síndrome de MEN 2B es una entidad clínica muy poco prevalente, pero altamente grave y con una elevada mortalidad. La dificultad para evaluar adecuadamente las características clínicas y los resultados de MEN 2B se relaciona con el bajo número de pacientes reportados en la literatura y el desconocimiento de esta enfermedad entre el personal de salud; esto explica por qué, a pesar de su primera descripción hace más de 50 años, la historia natural de MEN 2B sigue siendo imprecisa, lo que nos motiva a realizar esta revisión.
Desde el descubrimiento de las variantes genéticas se inició una búsqueda exhaustiva de asociaciones genotipo-fenotipo para comprender la alta variabilidad de presentación, con lo cual se ha logrado una mejor clasificación del riesgo y definir el pronóstico en relación con el genotipo, gracias a lo cual se ha podido mejorar el tratamiento de los pacientes mediante un asesoramiento genético exhaustivo. La sensibilización desde el punto de vista de asociaciones clínicas, la necesidad imperiosa de un estudio genético, el seguimiento de los familiares y la comprensión global del manejo óptimo temprano de los pacientes con MEN 2B permitirá en el futuro mejores desenlaces individuales y del grupo familiar que la padece.

