










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Universidad Industrial de Santander. Salud
Print version ISSN 0121-0807On-line version ISSN 2145-8464
Rev. Univ. Ind. Santander. Salud vol.42 no.1 Bucaramanga Jan./Apr. 2010
de un caso y revisión de la literatura
Albert Franz Guerrero, Carlos Augusto Cuadros, Diana Carolina Archila, Sandra Milena Beltrán,
Gustavo Adolfo Cuadros.
Departamento de Cirugía, Universidad Industrial de Santander, Bucaramanga, Colombia
Correspondencia: Albert Franz Guerrero. MD. Residente de Cirugía General III año. Carrera 8 - 61 Ciudadela Real de minas
Metrópolis II Torre 1 Apto 103 Bucaramanga. Colombia.
E-mail: albert_barza@hotmail.com
Recibido: 26 de noviembre de 2009 - Aceptado: 17 de diciembre de 2009
RESUMEN
Introducción: El síndrome de Prune Belly (SPB), también conocido como el síndrome de Eagle Barrett, se caracteriza por una triada de anomalías que incluye grados variables de hipoplasia de la musculatura abdominal, anomalías del tracto urinario y criptorquidia bilateral. Objetivo: Se describe el caso de un paciente masculino con Síndrome de Prune Belly y se realiza una revisión de la literatura sobre esta rara enfermedad. Conclusión: La característica arrugada del abdomen similar a una ciruela pasa, le da el nombre al síndrome. Además, puede estar asociado a alteraciones cardiovasculares, respiratorias, ortopédicas y gastrointestinales. Salud UIS 2010; 42: 78-85
Palabras Claves: Síndrome de abdomen en ciruela pasa, agenesia abdominal, anomalías del tracto urinario, criptorquidia
Prune Belly Syndrome: Case report and review
ABSTRACT
Introduction: Prune-belly syndrome, also known as Eagle-Barrett syndrome is characterized by a triad of anomalies that include varying degrees of abdominal musculature hypoplasia, urinary tract anomalies, and bilateral cryptorchidism. Objective: We describe the case of a male patient with Prune Belly Syndrome and we review the literature on this rare disease. Conclusions: The characteristic wrinkled, prune-like abdomen, gives the name to the syndrome. Can also be associated with cardiovascular, respiratory, orthopedic and gastrointestinal anomalies. Salud UIS 2010; 42: 78-85.
Keywords: Syndrome Prune-Belly, abdominal muscle deficiency, urinary tract abnormalities, cryptorchidism
INTRODUCCIÓN
El síndrome de Prune-Belly (SPB), también conocido como el síndrome de Eagle Barrett, se caracteriza por la triada que incluye diversos grados de hipoplasia de la musculatura abdominal, anomalías del tracto urinario y criptorquidia bilateral1,2. Hasta el 75% de los pacientes con SPB se han asociado con defectos a nivel pulmonar, cardíaco, óseo y gastrointestinal 3.
Forhlich en 1839 describió el primer caso de un niño que presentaba defecto de la musculatura abdominal, pecho en quilla y criptorquidia bilateral. En 1895 Parker asocia este síndrome con malformaciones de las vías urinarias: hidronefrosis, hidrouréter y megavejiga. El término "Prune Belly o abdomen en ciruela" fue acuñado por Osler en 1901, y refleja el aspecto arrugado de la piel de la pared abdominal del recién nacido debido a los diversos grados de hipoplasia de los músculos de la pared abdominal 4.
La incidencia de este síndrome se estima en 1 caso por cada 40.000 a 50.000 recién nacidos vivos, siendo mas frecuente en hombres que en mujeres, con una proporción de 18:1 (2). Por ser un síndrome infrecuente, se reporta el caso de un paciente masculino con Síndrome de Prune-Belly diagnosticado en la edad neonatal y se realiza una revisión de la literatura.
INFORME DEL CASO
Recién nacido pretérmino, masculino, de 2550 g, producto de madre primigestante de 18 años, la cual no realizó controles prenatales adecuados, quien fue hospitalizada en el Hospital Universitario de Santander (HUS) a las 33 semanas por preclampsia severa, síndrome de Hellp y oligoamnios, por lo cual se realizó cesárea de urgencia. Al nacimiento, el paciente presentó APGAR de 4 al minuto y 7 a los 5 minutos. Al examen físico se observó taquipneico, con dificultad respiratoria severa (Silverman Anderson 8 puntos). El examen pulmonar evidenció crépitos gruesos y tirajes subcostales. El abdomen era flácido, con aspecto arrugado. Adicionalmente los testículos no se localizaban en las bolsas escrotales. No se encontraron otros hallazgos relevantes.
Requirió intubación orotraqueal inmediata, junto con hospitalización en la Unidad de Cuidados Intensivos Pediátricos (UCIP) del HUS durante los primeros 12 días de vida debido a enfermedad de membrana hialina y neumonía nosocomial. Recibió soporte inotrópico, acompañado de ventilación mecánica por 7 días y tratamiento antibiótico de amplio espectro. Durante la estancia en UCIP por hallazgo de megalias bilaterales a nivel de flancos, de consistencia firme, se realizó una Ecografía abdominal que mostró hipoplasia renal derecha, hidronefrosis bilateral y megauréter de predominio izquierdo. La cistouretrografía miccional mostró reflujo vesicoureteral grado IV bilateral, sin obstrucción uretral. Pruebas de función renal: creatinina sérica: 0,7 mg/dl; nitrógeno ureico (BUN): 20mg/dl. Se realizó diagnóstico de SPB. Se descartaron defectos a nivel pulmonar, esquelético, cardíaco y gastrointestinal. Presentó buena evolución clínica, dando el alta hospitalaria a los 40 días de hospitalización.
Posteriormente requirió múltiples hospitalizaciones por síndrome broncoobstructivo y 2 episodios de infección urinaria; último episodio a los 2 años tratado con Ciprofloxacina por hallazgo de Escherichia coli resistente a Ceftriaxona. Se indicó profilaxis antibiótica con Ácido Nalidíxico. Además presentó detención del desarrollo psicomotor (solo se volteaba), estreñimiento crónico tratado con dieta rica en fibra y vejiga hiperactiva manejada con Oxibutinina. A los 2 años y 4 meses es llevado a cirugía para corrección de criptorquidia bilateral evidenciando criptorquidia derecha grado IV y anorquidia izquierda. Se realizó orquidopexia derecha.
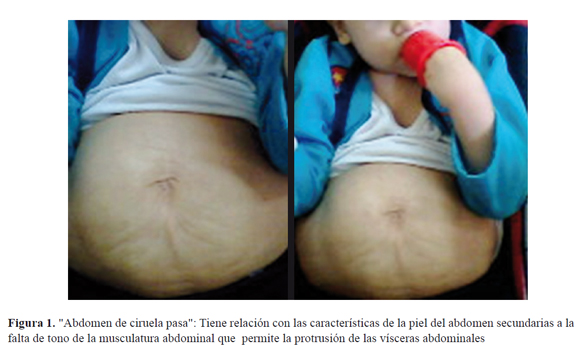
Actualmente el paciente tiene 3 años y 3 meses; no ha presentado nuevos episodios de infección urinaria, ni alteración de las pruebas de función renal. Continua manejo con profilaxis antibiótica con buena adherencia. Se encuentra en seguimiento pediátrico multidisciplinario. Presenta peso y talla en el percentil 50, cifras tensionales dentro del percentil para la edad. Abdomen con aspecto de ciruela pasa (Figura 1). Genitales con testículo derecho palpable en bolsa escrotal y testículo izquierdo ausente.
Laboratorios de control: creatinina sérica 0,6 mg/dl, BUN: 12 mg/dl, Parcial de orina normal, urocultivo negativo. Ecografía de vías urinarias de control muestra riñón derecho hipoplásico con corteza adelgazada, Hidronefrosis bilateral con predominio izquierdo y megauréter bilateral con predominio izquierdo, no progresivos. Cistouretrografía miccional muestra reflujo vesicoureteral bilateral no progresivo. Gammagrafía renal indica buena función y perfusión en el riñón izquierdo, con alteración de la función y perfusión del riñón derecho.
DISCUSIÓN
Se presenta el caso de un paciente preescolar con Síndrome de Prune-Belly diagnosticado en la edad neonatal, el cual presentaba la tríada característica: ausencia congénita de la musculatura de la pared abdominal; anormalidades del tracto urinario y criptorquidia bilateral. Los episodios de síndrome broncobstructivo, el estreñimiento y el retraso del desarrollo psicomotor que presentó el paciente podrían ser consecuencia de la hipoplasia de la musculatura de la pared abdominal. La hipoplasia del riñón derecho, hidronefrosis bilateral y reflujo vesicoureteral grado IV bilateral son factores de riesgo que podrían explicar los episodios de infección urinaria. Por esta razón se decidió llevar a cabo una profilaxis antibiótica indefinida con un riguroso seguimiento clínico, para evaluar la existencia de nuevos episodios de infección urinaria y llevar a cabo un tratamiento oportuno. Además, debido a que durante el procedimiento quirúrgico no se encontró el testículo derecho, el paciente debe continuar un seguimiento clínico, con el fin de establecer si realmente presenta anorquidia verdadera derecha, o si el testículo debido a su tamaño o localización no fue percibido durante la cirugía. De lo contrario aumentaría el riesgo de cáncer testicular.
Un factor que favoreció el pronóstico del paciente fue no presentar complicaciones diferentes a las que engloba la triada clásica de este síndrome, evitando la mortalidad generada por patología cardiopulmonar asociada y la morbilidad relacionada con la patología gastrointestinal y osteomuscular de este síndrome. Es importante resaltar que durante el embarazo la madre presentó oligohidramnios, el cual probablemente fue secundario a su patología de base no tratada, y pese a que el paciente tuvo complicaciones clínicas debidas a su prematurez, no presentó un marcado compromiso de la función renal, ni las manifestaciones extrarrenales del síndrome de Potter que hubieran empeorado su pronóstico.
Por otro lado, en el paciente no se constató la presencia de obstrucción uretral, por lo cual podríamos sugerir que este mecanismo patológico no fue la causa del SPB. Probablemente acorde a lo que argumentan muchos autores, la obstrucción uretral, más que ser la causa directa de la displasia renal, es un factor que contribuye a la progresión de esta.
En este momento el paciente se encuentra en manejo con profilaxis antibiótica por tiempo indefinido, tratamiento de la vejiga hiperactiva y seguimiento periódico de la función renal por un grupo multidisciplinario de pediatras, quienes definirán la necesidad de alguna intervención quirúrgica según su evolución, con el fin de evitar procedimientos invasivos innecesarios que predispongan a complicaciones que aumenten su morbimortalidad. La hipoplasia de musculatura abdominal y el retraso del desarrollo psicomotor, se han manejado de forma conservadora con terapia física.
Adicionalmente se debe resaltar que a pesar del reflujo vesicoureteral grado IV, la hipoplasia renal derecha y la hidronefrosis bilateral de este paciente; a lo largo del seguimiento no ha presentado clínica ni alteración de las pruebas de función renal, que indiquen la necesidad de diálisis o trasplante renal. El seguimiento clínico del paciente determinará si su único riñón funcional podrá mantener su buen resultado a lo largo de la infancia y la adolescencia.
Características clínicas
La piel laxa y arrugada de la pared abdominal, semejante a una ciruela pasa, es una de las principales características del SPB, causada por un defecto de la musculatura abdominal. Este defecto permite la visualización del peristaltismo intestinal y facilita la palpación de las estructuras intraabdominales y retroperitoneales1. La hipoplasia de la musculatura abdominal no sólo es una preocupación estética, también puede predisponer a otras alteraciones como retraso del desarrollo psicomotor, debido a que no permite mantener los niveles de fuerza requeridos para conservar el equilibrio y la estabilización de la columna, en la realización de diversas actividades como sentarse, caminar o correr5. Además, debido a que la musculatura abdominal participa en la realización de las maniobras de Valsalva, predispone a estreñimiento crónico y alteración del reflejo de la tos, generando evacuación inadecuada de cuerpos extraños de la vía aérea, que aumenta el riesgo de infecciones recurrentes del tracto respiratorio 2.
Las anormalidades del tracto urinario asociadas con SPB incluyen dilatación de diversas estructuras, tales como megavejiga, megauréteres e hidronefrosis, las cuales predisponen a alteraciones de la micción, reflujo vesicoureteral, estasis, infecciones recurrentes del tracto urinario e hipoperfusión renal2, 6. Una característica del SPB en los hombres es la marcada dilatación de la uretra prostática que conduce a hipoplasia o ausencia de tejido prostático, causada por una implantación anormal de la uretra membranosa sobre la uretra prostática. Debido a esto en ocasiones desarrollan un mecanismo de válvula que causa obstrucción urinaria, el cual simula la presencia de valvas uretrales2.
Los hombres con SPB presentan criptorquidia bilateral, frecuentemente localizados dentro de la pared posterior de la cavidad abdominal. Hasta el momento no existen informes que confirmen casos de fertilidad, pese a la realización de orquidopexia temprana7. Los niveles de testosterona, la libido y la función sexual son normales. En mujeres se ha descrito un pseudosíndrome equivalente, caracterizado por hipoplasia de la musculatura abdominal, alteraciones congénitas del aparato urinario y anomalías genitales tales como útero bicorne y atresia vaginal, fístula vesico-vaginal, anomalías del seno urogenital y anomalías ano-rectales; el cual se diferencia del SPB por la ausencia de obstrucción uretral7.
Además de las anormalidades descritas, este síndrome se ha asociado con otras alteraciones. La mayoría de los pacientes presentan grados variables de hipoplasia pulmonar, resultado del oligohidramnios generado a causa de la displasia renal. Esta puede verse agravada por anomalías esqueléticas de la caja torácica y la alteración de la musculatura abdominal1,2. Estos pacientes son propensos a desarrollar infecciones respiratorias recurrentes8,9. Además se han descrito deformidades esqueléticas hasta en el 45% de los pacientes con PBS; también secundarias al oligohidramnios, incluyen pie zambo, luxación congénita de la cadera, ausencia de las extremidades, pectus excavatum, y escoliosis10. Las anomalías cardíacas como persistencia del conducto arterioso, comunicación interauricular, defecto septal ventricular y la tetralogía de Fallot, se han informado hasta en el 10% de los pacientes1,2. También se ha asociado con alteraciones gastrointestinales como mal rotación intestinal, atresia anal, estenosis de intestino delgado y vólvulos. El estreñimiento crónico, se encuentra en más del 30% de pacientes2. El retraso del crecimiento y desarrollo también se ha asociado con este síndrome2,10.
Etiología
Aunque se desconoce la causa del SPB, actualmente dos teorías se destacan. Una teoría propone que las anomalías asociadas al SPB son el resultado de un desarrollo anormal de la placa intermedio-lateral del mesodermo. Un defecto primario a este nivel afectaría la embriogénesis de la musculatura de la pared abdominal, los conductos mesonéfricos y paramesonéfricos, y los órganos urinarios11. Otra teoría plantea la existencia de una obstrucción a nivel distal de la uretra en una edad gestacional temprana, la cual provoca dilatación masiva de la vejiga y los uréteres, ejerciendo presión sobre la musculatura abdominal y causando un efecto de barrera física que dificulta el desarrollo de musculatura de la pared abdominal, de la próstata; y el descenso de los testículos12. Algunos autores sugieren que estas teorías deben ser consideradas como complementarias7.
Clasificación
Una clasificación ampliamente aceptada debido a su valor pronóstico, subdivide los pacientes en tres grupos basados en el estado del tracto urinario durante la infancia13. (Tabla 1). Los niños más afectados son los que tienen obstrucción uretral completa, displasia renal grave y función renal mínima. Estos pacientes, a menudo tienen las manifestaciones extrarrenales del síndrome de Potter, incluyendo la hipoplasia pulmonar13. Pueden nacer muertos o morir poco después del nacimiento debido a insuficiencia pulmonar y / o renal. Entre los pacientes que sobreviven el período neonatal inmediato, 30% desarrollan insuficiencia renal crónica en estadio terminal que requiere diálisis o trasplante durante la infancia o adolescencia2. Los pacientes con menor afectación del tracto urinario, sin obstrucción ni alteración de la función renal, tienen una expectativa similar a la población sin esta afección13. La tasa de mortalidad general en SPB es del 60%14.

Diagnóstico
El diagnóstico del SPB puede realizarse durante la gestación a través de la ecografía obstétrica la cual puede evidenciar aumento de la circunferencia abdominal con contornos irregulares, debido a la ausencia de la pared muscular abdominal; riñones poliquísticos o displásicos; hidroureteronefrosis; dilatación de uréteres y vejiga; oligohidramnios; ascitis fetal y uraco permeable15. La ecografía transvaginal de alta resolución permite la visualización de los riñones y el tracto urinario desde la semana 11 a 13 de gestación 15. De ahí la importancia de un diagnóstico prenatal ya que permite una intervención oportuna del niño, informa con anticipación a los padres de esta malformación, de modo que se preparen emocionalmente para recibir a un hijo con esas condiciones. Además es una herramienta que permite al equipo médico planificar un trabajo interdisciplinario y coordinado entre obstetras, neonatólogos, nefrólogos y cirujanos pediatras, fundamental para la sobrevida y pronóstico de los niños que sufren este tipo de malformaciones4.
Sin embargo, este síndrome se diagnostica con mayor frecuencia durante el periodo neonatal, por la particular apariencia de la piel de la pared abdominal. Existen casos en los cuales no hay signos clínicos sugestivos de abdomen en ciruela pasa; en ellos, el diagnóstico se hace a edades más tardías y requiere un alto índice de sospecha clínica16,17.
El objetivo de la evaluación postnatal es identificar las enfermedades que amenazan la vida del recién nacido, por lo tanto primero se debe excluir posible patología cardiaca y pulmonar1,2. Posteriormente el paciente debe someterse a una evaluación del tracto urinario, realizando pruebas de laboratorio como nitrógeno ureico plasmático y creatinina sérica para evaluar la función renal, parcial de orina y urocultivo para descartar infección urinaria; y la obtención de imágenes diagnósticas, con el fin de evaluar la anatomía renal, descartar procesos infecciosos de parénquima renal y justipreciar la función renal1, 2. La ecografía de vías urinarias permite observar la anatomía del riñón, uréteres y vejiga. La cistouretrografía miccional no era realizada de rutina, por ser un estudio invasivo que se asociaba con riesgo de infección urinaria18, no obstante, recientemente es llevada a cabo para descartar patología obstructiva. Estudios adicionales, como la gammagrafía renal y urodinamia, pueden ser necesarios para definir mejor la anatomía y fisiología renal. Debido a las implicaciones pronósticas de afectación del tracto urinario, los pacientes deben ser evaluados con frecuencia, con el propósito de evaluar la posibilidad de infección del tracto urinario, disfunción vesical e insuficiencia renal1, 2.
Tratamiento
El tratamiento del recién nacido con SPB tiene como objetivo prevenir el deterioro de la función renal. Intervenciones quirúrgicas tempranas no justificadas pueden empeorar el pronóstico de estos pacientes, por lo tanto, se prefiere realizar un manejo conservador, llevando a cabo una evaluación minuciosa de la función renal durante la primera semana de vida. En caso de evidencia de reflujo vesicoureteral severo, asociado a insuficiencia renal o hidronefrosis severa, se deben corregir las anormalidades bioquímicas e instaurar profilaxis antibiótica para evitar infecciones secundarias. Algunos casos requieren ureterostomías cutáneas de emergencia una vez se han corregido las alteraciones bioquímicas y la infección, para lograr disminuir la presión del sistema urinario y mejorar la función renal 19.
El tratamiento del SPB en la primera infancia sigue siendo controvertido. Los pacientes con reflujo vesicoureteral severo requieren cirugía de drenaje urinario (nefrostomía, ureterostomía o cistostomía supravesical), las cuales pueden proporcionar un mejor drenaje urinario y evitar infecciones recurrentes20. En estos casos el uso de antibióticos profilácticos durante tiempo indefinido esta indicado con el fin de prevenir las infecciones recurrentes del tracto urinario y pielonefritis21. La Cistoplastia de reducción puede ser utilizada en pacientes con incontinencia urinaria, para mejorar la función contráctil del músculo detrusor y disminuir el riesgo de infección urinaria22. La hemodiálisis o diálisis peritoneal continua, están indicadas en pacientes con insuficiencia renal terminal. Ambas tienen igual eficacia en el manejo de la enfermedad renal terminal 23. El Trasplante renal en estos pacientes ha mostrado excelentes resultados a largo plazo24.
La hipoplasia de la musculatura abdominal no tiene repercusión sobre el pronóstico de estos pacientes, a pesar de las alteraciones que produce sobre la dinámica respiratoria. Con el paso de los años, hay un aumento de la grasa subcutánea a este nivel, que mejora el tono de la pared abdominal. Por esta razón, rara vez se realizan cirugías reconstructivas de pared abdominal. Sin embargo, la pared abdominal puede ser reconstruida por razones estéticas y psicosociales, por vía intraperitoneal (plicatura de Monfort) o vía extraperitoneal por laparoscopia26,27. Además de mejorar el aspecto abdominal, estas técnicas también aumentan la dinámica respiratoria y el vaciamiento vesical e intestinal26. Adicionalmente pueden reducir las deformidades esqueléticas, evitando la pérdida de estabilidad de la columna y el desarrollo de cifoescoliosis28.
La orquidopexia temprana es importante en los niños con SPB, ya que previene la aparición de malignidad y preserva de cierta forma la fertilidad29. Un estudio retrospectivo de 41 hombres adultos con SPB, a quienes se les realizó orquidopexia durante la infancia, reveló que el 93% de pacientes lograron una función testicular óptima para inducir la pubertad y mantener una función sexual satisfactoria en la vida adulta30.
CONCLUSIÓN
El SPB es una enfermedad congénita poco frecuente compuesta por anomalías de varios órganos. Tiene un amplio espectro clínico que va desde los niños gravemente comprometidos con riesgo de morir desde el nacimiento, hasta los que crecen normalmente con una función renal satisfactoria. El tratamiento de esta enfermedad varía en función de la severidad del caso. Es importante para los médicos que participen en el cuidado de estos pacientes estar familiarizados con el manejo y las posibles complicaciones que se puedan presentar.
AGRADECIMIENTOS
Al Hospital Universitario de Santander en donde se llevó a cabo el estudio de este caso.
CONSIDERACIONES ÉTICAS
Los padres del paciente incluido en este estudio firmaron de manera libre y voluntaria un consentimiento informado construido acorde con la Resolución 008430/93 dictaminada por el Ministerio de Salud de Colombia. Se aseguró absoluta reserva de la identidad del paciente.
CONFLICTO DE INTERÉS
Los autores declaran no tener ningún conflicto de intereses en la realización del presente trabajo.
REFERENCIAS
1. Diao B, Diallo Y, Fall PA, Ngom G, Fall B, Ndoye AK, Fall I, Ba M, Ndoye M. Prune Belly syndrome: Epidemiologic, clinic and therapeutic aspects. Prog Urol 2008; 18: 470-474. [ Links ]
2. Woods Amanda, Brandon Debra. Prune Belly Syndrome: A Focused Physical Assessment. Advances in Neonatal Care 2007; 7: 132-143 [ Links ]
3. Denes FT, Arap MA, Giron AM, Silva FAQ, Arap S. Comprehensive surgical treatment of prune belly syndrome: 17 years' experience with 32 patients. Urology 2004; 64:789-793. [ Links ]
4. Wallner Manfred, Kramar Reinhard, Prune-belly síndrome. UpToDate. Last literature review version 17.2: may 2009. [ Links ]
5. Cissik J.M., Programming Abdominal Training, Part I. Journal of Strength and Conditioning 2002; 24:9-15 [ Links ]
6. Reinberg, Y, Manivel, JC, Pettinato, G, et al. Development of renal failure in children with the prune belly syndrome. J Urol 1991; 145:1017-1019. [ Links ]
7. Reinberg, Y, Shapiro, E, Manivel, JC, et al. Prune belly syndrome in females: A triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr 1991; 118:395-398. [ Links ]
8. Crompton CH, MacLusky IB, Geary DF. Respiratory function in the prune-belly syndrome. Arch Dis Child 1993; 68:505–506. [ Links ]
9. Ewig, JM, Griscom, NT, Wohl, ME. The effect of the absence of abdominal muscles on pulmonary function and exercise. Am J Respir Crit Care Med 1996; 153:1314-1321. [ Links ]
10. Loder RT, Guiboux JP, Bloom DA et al. Musculoskeletal aspects of prune-belly syndrome. Am J Dis Child 1992; 146:1224–1229. [ Links ]
11. Stephens, FD, Gupta, D. Pathogenesis of the prune belly syndrome. J Urol 1994; 152:2328-2331. [ Links ]
12. Volmar, KE, Fritsch, MK, Perlman, EJ, Hutchins, GM. Patterns of congenital lower urinary tract obstructive uropathy: relation to abnormal prostate and bladder development and the prune belly syndrome. Pediatr Dev Pathol 2001; 4: 467-472. [ Links ]
13. Loredo R, Fernández SA., Ochúa G M., Carpio RA. Complejo malformación-obstrucción uretral: diagnóstico ecográfico prenatal. Revista Argentina de Radiología. 2008; 72:439-442 [ Links ]
14. Druschel CM. A descriptive study of prune belly in New York State, 1983 to 1989. Arch Pediatr Adolesc Med 1995; 149:70–76. [ Links ]
15. Yamamoto, H, Nishikawa, S, Hayashi, T, et al. Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res 2001; 27:37-40. [ Links ]
16. Wallner, M, Kramar, R. Detection of prune-belly syndrome in a 35-year-old man: A rare cause of end-stage renal failure in the adult. Am J Nephrol 1990; 10:413-415. [ Links ]
17. Brown, PS, Hartz, RS, Hines, JR. Prune belly syndrome in adult man. Urology 1993; 41:199. [ Links ]
18. Smith E, Woodard J. Prune-belly syndrome. Campbell's urology. Eighth Edition. Philadelphia WB Saunders 2002; 2117-2133. [ Links ]
19. Woodard, JR, Zucker, I. Current management of the dilated urinary tract in prune belly syndrome. Urol Clin North Am 1990; 17:407-418. [ Links ]
20. Milliken, I, Munro, NP, Subramaniam, R. Cystostomy button for bladder drainage in children. J Urol 2007; 178:2604-2606. [ Links ]
21. Woodhouse, CR, Neild, GH. A child with recurrent urine infections and undescended testes. Nephrol Dial Transplant 1995; 10:711-713. [ Links ]
22. Bukowski, TP, Perlmutter, AD. Reduction cystoplasty in the prune belly syndrome: A long-term followup. J Urol 1994; 152:2113-2116. [ Links ]
23. Wisanuyotin, S, Dell, KM, Vogt, BA, et al. Complications of peritoneal dialysis in children with Eagle-Barrett syndrome. Pediatr Nephrol 2003; 18:159-163. [ Links ]
24. Fusaro, F, Zanon, GF, Ferreli, AM, et al. Renal transplantation in prune-belly syndrome. Transpl Int 2004; 17: 549-552. [ Links ]
25. Kamel, MH, Thomas, AA, Al-Mufarrej, FM, et al. Deceased-donor kidney transplantation in prune belly syndrome. Urology 2007; 69: 666-669. [ Links ]
26. Furness PD, 3rd, Cheng, EY, Franco, I, Firlit, CF. The prune-belly syndrome: a new and simplified technique of abdominal wall reconstruction. J Urol 1998; 160:1195-1197. [ Links ]
27. Franco, I. Laparoscopic assisted modification of the firlit abdominal wall plication. J Urol 2005; 174: 280-283. [ Links ]
28. Lam, KS, Mehdian, H. The importance of an intact abdominal musculature mechanism in maintaining spinal sagittal balance. Case illustration in prune-belly syndrome. Spine 1999; 24: 719-722. [ Links ]
29. Redman, JF, Mooney, DK. Fowler-Stephens orchiopexy in a patient with prune belly syndrome and segmental atretic vas deferens. Urology 1993; 41: 130-131. [ Links ]
30. Patil, KK, Duffy, PG, Woodhouse, CR, Ransley, PG. Long-term outcome of Fowler-Stephens orchiopexy in boys with prune-belly syndrome. J Urol 2004; 171: 1666-1669. [ Links ]

