











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
The SARS-CoV-2 virus, the causal agent of the coronavirus disease COVID-19, was first identified at the end of 2019 in Wuhan, China 1. Given its transmission via the respiratory tract, the global infection rate became increasingly high, leading the World Health Organization [WHO] to declare a pandemic in 2020 2. To curb its spread, the WHO recommended diagnosing this disease using purified RNA from nasopharyngeal swabs through a One-Step Reverse Transcription Quantitative Real-time PCR [RT-qPCR]. For this purpose, diverse primers and probes for RT-qPCR were designed supported on the initial characterization of the SARS-CoV-2 genome, which is a positive-sense single-stranded RNA of approximately 30 kb, containing 10 genes 1,3. The Charité-Berlin protocol, which is widely recommended by the WHO, targets regions of the genes coding for RNA-dependent RNA polymerase [RdRp], Envelope protein [E] and Nucleocapsid phosphoprotein (N) 4. Another diagnostic kit, designed by the Centers for Disease Control and Prevention-USA [CDC], utilizes two regions of the N gene and has proven successful in occupational medicine for singleplex and/or multiplex RT-qPCR to single and/or pooled nasopharyngeal swab samples 5,6. Meanwhile other diagnostic kits use regions of the Spike (S) gene 7.
Despite the widespread application and accuracy of RT-qPCR for SARS-CoV-2 detection, previous studies on other RT-PCR methods have reported that the procedure may yield false negative results or low-quality amplification signals, leading to incorrect viral sample detection 8. This is due to alterations in the hybridization pattern between the primers or probes caused by single nucleotide polymorphism in the target regions of the viral variants 9,10and/or the polymerization capacity of DNA polymerase affected by the formation of RNA or DNA secondary structures that regulate gene expression 11.
A notable feature of the SARS-CoV-2 genome is the folding at the 5' and 3' genome untranslated regions, creating secondary stem-loops structures involved in regulating the translation of the first gene and the genome's half-life, respectively 12. Additionally, certain viral RNA stems induce Programmed Ribosomal Frameshift in the ORF1a and ORF1b genes, altering translation at the stop codon to generate ORF1ab, a component of the viral RNA polymerase 13.
To address the challenges associated with RNA structures, we implemented a Denaturing Solution (DS) to prevent RNA folding in RT-qPCR, which contains Tetramethylammonium Chloride (TMAC) and Dimethyl Sulfoxide (DMSO) due to their affinity to A-T and G-C canonical bonds, respectively 14. In turn, the concentration of this DS was adjusted in accordance with the nucleotide composition of the SARS-CoV-2 genome, which is rich in A-T1. As a result, the DS facilitates the elimination of viral RNA secondary structures that could interfere with proper polymerization 15.
This innovative formulation was then incorporated into RT-qPCR protocol for the examination of nasopharyngeal swab samples, yielding an improved polymerization signal or multiplex for SARS-CoV-2 detection 15. However, many patients found nasopharyngeal swab sampling inconvenient, and it is also resource-consuming given the skilled staff required for the procedure 16,17. Then, implementing this approach for a large number of patients proved complex and costly 18,19. In response to these limitations, procedures involving individual or pooled saliva samples were introduced 20, the effect of the DS on these procedures in individual or pooled RT-qPCR in saliva pools had not been assessed, therefore, the objective of this study was to demonstrate that including the DS in RT-qPCR for SARS-CoV-2 detection, using N1 and N2 primers and probes for their respective regions in the N gene, enhances the polymerization signal in saliva samples collected over a 6-month period in Bucaramanga, Colombia. Taken together, our findings are expected to contribute to a better interpretation of viral detection.
Methodology
Ethics Statement
This study was approved by the Comité de Ética en Investigación Científica de la Universidad Industrial de Santander. Furthermore, this research adhered to all bioethical standards of the Declaration of Helsinki, revised in 2013 21.
Homology pattern to primers/probes N1-N2 regions of N gene
Consensus genomic sequences for SARS-CoV-2 variants Alpha, Beta, Delta, Gamma, GH490R, Lambda, Mu, and Omicron from the year 2021 were obtained from the public database of SARS-CoV-2 from Cadena Caballero et al., 2023 22, with nucleotide frequency thresholds of 20% and 100%. Alignments with respect to the SARS-CoV-2 reference sequence 23 were conducted using the MEGA software version ll with default parameter 24. Afterward, the percentage of identity for Nl and N2 primers and probes from the CDC SARS-CoV-2 protocol 6 [2019-nCoV TaqMan RT-PCR Kit - Norgen Biotek Corp.], was calculated for the N1 and N2 regions from the N gene reference and SARS-CoV-2 variant genes in terms of nucleotide changes per position, as indicated by the Nomenclature Committee of the International Union of Biochemistry (NC-IUB):
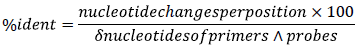
The total nucleotide combinations by position in the NC-IUB for N1 and N2 primers and probes of the N gene in SARS-CoV-2 variants were determined by constructing a matrix in a spreadsheet.
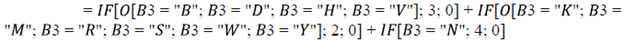
Where the number of nucleotide combinations was indicated by: K, M, R, S, W or Y = 2; B, D, H or V = 3; and N = 4.
RNA structures from N1-N2 regions of the N gene
The patterns of stem and stem-loop RNA secondary structures of N1, N2, and RNase P regions of the respective genes from the CDC detection kit 6 were generated in Mfold software version 2.3 with default parameters 25. VARNA version 3.93 was used to visualize the RNA secondary structures and RNAcomposer version 1.0 was used to obtain the RNA tertiary structures 26,27.
RNA extraction from saliva samples
Saliva samples, including 40 positive and 10 negative samples of SARS-CoV-2, were donated by Laboratorio Central de Investigaciones de la Facultad de Salud de la Universidad Industrial de Santander, collected by this institution from January to June 2022. For each sample, cell lysis, and purification of total RNA was carried out from 200 JL, using mechanical disruption and separation with magnetic beads with the MagMAX™ Viral/Pathogen II kit [MVP II -2000 RXN - Applied Biosystems - USA] using the software BindIt version 4 on the equipment the KingFisher Duo Prime [Catalog number: 5400110 - DNA/RNA extraction system] according to the manufacturer's instructions (Thermo Fisher Scientific - USA).
Denaturing Solution (DS) in individual and pooled RT-qPCR
The effect of DS in singleplex RT-qPCR for SARS-CoV-2 single saliva samples was assessed using the 2019-nCoV TaqMan RT-PCR Kit protocol (Ref. TM67100, Norgen Biotek - Canada) 6. Total RNA extractions from 20 of the 40 positive samples were randomly selected, and two sets of reactions were conducted. The first set included DS, and each reaction was conducted in a 15 volume composed by: 2 of total RNA; 7.5 μL of 2x One-Step RT-PCR Master Mix; 1 μL of DS (636 mM TMAC - ABCAM USA) and 423 mM DMSO (Scharlab - Spain] dissolved in nuclease-free water to a final reaction concentration of 21.2 mM and 14.1 mM, respectively) 22; 1.2 μL 2019-nCoV Primer & Probe Mix to N1, N2 or RNase P (per sample); and 3.3 nuclease-free water. The second reaction set did not include DS because it was controlled, therefore 4.3 μL nuclease-free water was used.
The One-Step RT-qPCR cycling conditions consisted of: first, a step at 50 °C for 30 minutes; second, a step at 95 °C for 3 minutes; third, 45 cycles distributed in two steps, one at 95 °C for 3 seconds and the other at 55 °C for 30 seconds. For this, a QuantStudio 1 real-time PCR system (No. A40427) was used in a 96-well block of 0.2μL (Thermo Fisher Scientific, USA).
For multiplex RT-qPCR to pooled samples, total RNA was obtained following the previous procedure from 11 pools, each comprising 200 μL from 10 saliva samples randomly selected from the initial 50 samples. To generate each pool, 20 μL by negative (n-) and positive (n+) SARS-CoV-2 samples were mixed in a proportion of: 10 negative samples and 0 positive; 9 negative and one positive; and so on until 10 positive samples and 0 negative. The RT-qPCR set for each pool included the standard procedure: 2 μL of RNA sample mix from each pool, 7.5 μL of 2x One-Step RT-PCR Master Mix; 1 of DS as previous indicated; 1.2 μL from 2019-nCoV Primer & Probe Mix to N1 and the same volume to N2 Primer & Probe Mix; and 3.3 μL nuclease-free water. The second RT-qPCR set served as the control, so did not include DS and 4.3 μL of nuclease-free water was used instead. The same procedure was applied to RNase P serving as the RT-qPCR control. The RT-qPCR Program was conducted as described above.
Statistical analysis
Box plot diagrams were generated using the Cycle threshold (Ct) distribution from each individual and pooled RT-qPCR set with and without DS, indicated as standard. Furthermore, a t-test was conducted to establish the statistically significant difference between the Ct average of the individual RT-qPCR with and without DS. Shapiro-Wilk and Levene's tests were also performed to evaluate the normality of the data and the homogeneity of the variances, respectively. In cases where the results did not exhibit a normal distribution, Wilcoxon-Mann-Whitney test (U-Test) was employed for group comparison between the Ct values groups. All statistical analyses were conducted in RStudio version 2023.03.0+386 and R version 4.2.2 28,29.
Results
Homology pattern of NI-N2 primers/probes
At the 20% frequency threshold, consensus sequences for N1 in the variants Alpha, Beta, Delta 1, Delta 2, Gamma, and Mu, exhibited 100% identity with the corresponding primers and probes sequences recommended by the CDC for SARS-CoV-2 detection via RT-qPCR. However, for GH490R, Lambda, and Omicron, the identity exceeded 97%. In contrast, the primer and probe sequences for the N2 region showed consistent identity across all SARS-CoV-2 variants. The consensus sequences with a 100% frequency threshold for primers and probes to the N1 region of Gamma, GH490R, Lambda and Omicron variants exhibited an average identity of 96%, Beta and Mu variants 72.1% on average, while Delta 1 and 2 showed the least identity (Table 1). The N1 primers and probes regions of the GH490R variant with a 20% frequency threshold, showed a nucleotide pair combination, whereas the Lambda and Omicron variants displayed a single change, according to NC-IUB nucleotide nomenclature. For other N1 regions in SARS-CoV-2 variants, including the N2 region, nucleotide changes were not determined (Table 1).
Table 1 Identity percentages and number of potential nucleotide changes for the SARS-CoV-2 variants from 2021 in N1 and N2 regions of N gene.
| N1 | N2 | |||||||
|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2 variant consensus | Threshold frequency 20% | Threshold frequency 100% | Threshold frequency 20% | Threshold frequency 100% | ||||
| ID % | N1 Changes | ID % | N1 Changes | ID % | N2 Changes | ID % | N2 Changes | |
| Alpha | 100 | 0 | 60.3 | 32 | 100 | 0 | 75.4 | 17 |
| Beta | 100 | 0 | 70.6 | 22 | 100 | 0 | 88.5 | 8 |
| Deltal | 100 | 0 | 45.6 | 48 | 100 | 0 | 60.7 | 29 |
| Delta2 | 100 | 0 | 32.4 | 68 | 100 | 0 | 52.5 | 40 |
| Gamma | 100 | 0 | 98.5 | 1 | 100 | 0 | 100 | 0 |
| GH490R | 97.1 | 2 | 97.1 | 2 | 100 | 0 | 100 | 0 |
| Lambda | 98.5 | 1 | 92.6 | 5 | 100 | 0 | 98.4 | 1 |
| Mu | 100 | 0 | 73.5 | 20 | 100 | 0 | 78.7 | 13 |
Note: Threshold frequency 20-100%: indicates the threshold employed to obtain consensus sequences for the SARS-CoV-2 variants sequences. ID %: indicates the percentage of identity for primers and probes designed to detect N1 and N2 region of N gene, for consensus sequences of SARS-CoV-2 variants. Nl-N2 Changes: indicates the number of possible nucleotide combinations per position for the N1 and N2 region of N gene, of the SARS-CoV-2 variants consensus sequences.
For SARS-CoV-2 variants with a 100% frequency threshold in primers and probes for the N1 region, Delta l and 2 variants showed the most significant changes per position. Alpha and Beta variants showed 50.5 changes in average, and except for the Mu variant, the other nucleotide combinations in the consensus regions of the SARS-CoV-2 variants exhibited fewer than 10 changes per position (Suppl. Material l). In the N2 region, the nucleotide combinations for all variants were similar to the N1 region, but the total number of combinations was lower. The Delta 1 and 2 variants had more than 100 nucleotide combinations, different for Gamma, GH490R, and Omicron variants, where the identity was the same as the reference sequence; while Alpha, Beta, and Mu showed an average of 24.3 nucleotide combinations (Suppl. Material 1).
Secondary and tertiary RNA structures for NI-N2 of the N gene
The secondary structures from the N1 region to all SARS-CoV-2 variants with a 20% frequency threshold generated the same structure, consisting of two stem-loop structures separated by 14 nucleotides. Notably, nucleotide combinations determined for GH490R were localized in the first loop of the first stem-loop and the region separating both stem-loops. Meanwhile Lambda and Omicron variants exhibited a T-C variation in the same nucleotide position of the second loop from the first stem-loop. A similar RNA secondary structure was observed in the N2 region and the RNase P sequence amplicon, but the stem-loop pairs were continuous in both cases (Figure 1).
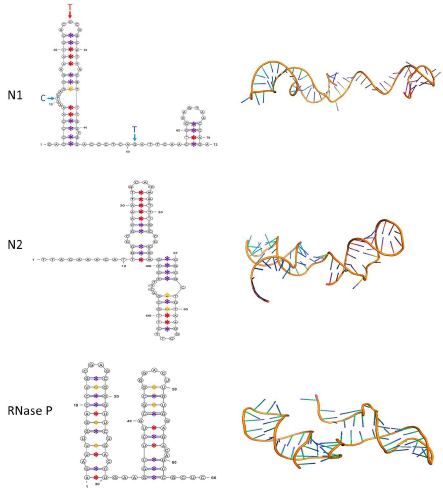
Fig. 1 Secondary and tertiary structures of the N1 and N2 regions of the SARS-CoV-2 N gene (NC_045512, Region: 28274-29533, position 14-86 and 891-957, respectively) and human RNase P. In the secondary structures of each of the amplicons (left), the nucleotide changes of the variant GH490R are indicated by blue letters and arrows, whereas the red arrow corresponds to Lambda and Omicron. Canonical A-T and G-C base pairings are shown with red and purple asterisks, respectively. Non-canonical Guanine-Thymine (G-T) pairings are marked with yellow asterisks. The tertiary structures of each region are displayed on the right
It is also worth noting that in the secondary structure of N1 and N2 regions of SARS-CoV-2 variants, the number of G-C base pairs in all stem-loop structures was determined at the beginning of each stem-loop. In contrast, A-T and non-canonical base pairs were preferably localized in the superior region of the stem-loop. In the RNase P stem-loops, the base pairs did not follow a specific pattern distribution (Figure 1).
DS improves SARS-CoV-2 detection by individual RT-qPCR
In the individual RT-qPCR for the N1 region, the expected result was obtained in 18 out of 20 samples, even after the addition of the DS. Moreover, amplifications for the N2 region and RNase P were successful in all samples with both treatments (Figure 2A). The average Ct values obtained for individual RT-qPCRs with DS and the standard solution showed differences for the N1 and N2 regions of the SARS-CoV-2 N gene and RNase P. With the inclusion of DS in single RT-qPCR for the N1 region and RNase P the Ct average values were lower compared to the standard reaction. However, a slight increase in Ct average value was determined in presence of DS when targeting the N2 region via RT-qPCR (Figure 2A).
The Shapiro-Wilk's normality test revealed that the Ct values for individual RT-qPCR N1-DS (Ct mean= 21.48, SD= 8.44), N1-Standard (Ct mean= 22.32, SD = 9.18), and RNase P-Standard (Ct mean= 22.65, SD= 2.07), did not follow a normal distribution. While the RT-qPCR of N2-DS (Ct mean= 23.82, SD= 5.19), N2-Standard (Ct mean= 23.21, SD= 4.73), and RNase P-DS (Ct mean= 21.88, SD= 1.72) Ct values exhibited a normal distribution. Furthermore, all RT-qPCR treatments had homogenous variances according to Levene's Test. However, according to our t-test analyses, there were no statistically significant differences between the Ct averages of the N2-DS and N2-Standard RT-qPCR. For groups without a normal distribution, U-Test results also showed no differences between the Ct average values (Table 2).
Table 2 The t-test and U-test results indicate no statistically significant differences between the Ct averages, p-value= 0.05.
| Individual RT-qPCR | Shapiro-Wilk's normality test | Levene's Test | T-Test | Wilcoxon-Mann-Whitney test (U-Test) |
|---|---|---|---|---|
| N1-DS | 0.001484* | 0.7618 | N.D | 0.735 |
| N1-Standard | 0.01227* | |||
| N2-DS | 0.3507 | 0.5421 | 0.698 | N.D |
| N2-Standard | 0.604 | |||
| RNase P-DS | 0.6378 | 0.8897 | N.D | 0.2012 |
| RNase P-Standard | 0.0415* |
Note: DS indicates RT-qPCR reactions with the Denaturant Solution. Standard indicates RT-qPCR reactions without the Denaturant Solution. Values with *, indicates Ct average without a normal distribution. N.D indicates non-determined test according to non-compliance their assumptions.
DS improve SARS-CoV-2 detection by pooled RT-qPCR
The pooled RT-qPCR for the Nl/N2 regions in each of the ll pooled samples and RNase P produced the expected result. The Ct average for N1/N2 regions and RNase P showed a decrease with the DS solution compared to the standard solution (Figure 2B). Notably, the onset of the Log phase in the RT-qPCRs was directly proportional to the number of positive samples within each pooled sample, and the number of cycles required to obtain their Ct was lower when DS was included in each pooled RT-qPCR. Moreover, the inclusion of DS showed a remarkable difference in the fluorescence signal, being higher than the signal obtained in the standard RT-qPCR (Figure 3).
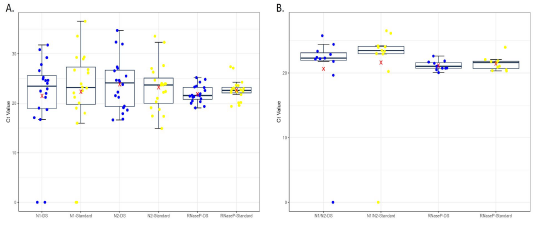
Fig. 2 Boxplot of Ct values from single (A) and pooled (B) RT-qPCR of the Nl and N2 regions of the SARS-CoV-2 N gene and human RNase P reaction set. The Ct average for each RT-qPCR set is marked with a red X. CT's value from sets with DS in the RT-qPCR are represented by blue dots, while the control set is indicated by yellow dots.
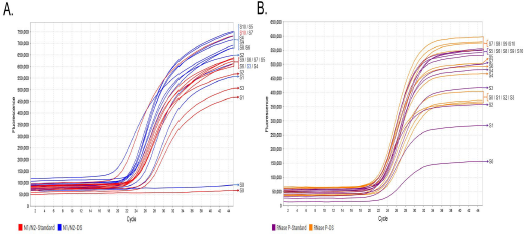
Fig. 3 Multiplex RT-qPCR for the characterization of the N gene and RNase P from SARS-CoV-2 using pooled saliva samples. The fluorescence signals emitted by the Nl/N2 primers and probes targeting the respective SARS-CoV-2 N gene with DS are represented by blue curves, whereas the control RT-qPCR is indicated by red curves (A); and RNase P with DS are represented by orange curves, whereas the control RT-qPCR is indicated by purple curves (B). The numbers on the right side of the curve in both graphs indicate the number of positive SARS-CoV-2 RNA samples relative to the negative samples in the corresponding pool.
Discussion
Although RT-qPCR is widely regarded as the gold standard for SARS-CoV-2 diagnosis, it may occasionally yield false-negative results due to mutational patterns in new variants 30 and the secondary structures of viral RNA 12. In this sense, the determination of those structures continues to be a challenge to establish its therapeutic potential and their elimination from RNA/DNA polymerase processes31; as demonstrated in the Hepatitis Delta Virus32 or the bacterium Klebsiella pneumoniae33. The findings from this study indicate that implementing DS to prevent RNA folding in RT-qPCR from saliva samples enhances the fluorescent signal and Ct values, both in single samples and multiplex pooled samples, for the N1 and N2 regions of the N gene used in the CDC protocol 6.
In silico analysis conducted in this study on some medically relevant SARS-CoV-2 variants during the COVID-19 pandemic previously reported 15, underscores the importance of comparing the identity between primers and probes respect to consensus regions employed in RT-qPCR for virus detection. In addition, the threshold frequency allows to determine the nucleotide polymorphism in a specific region of a gene or genome, which can be visualized in a consensus sequence based on the threshold frequency value 34. In this study, the consensus regions obtained with a 20% threshold demonstrated a correct identity pattern for the N1 region in Alpha, Beta, Delta, Gamma, and Mu SARS-CoV-2 variants, while the N2 region showed complete identity across all eight variants. However, the N1 region of the GH490R, Lambda, and Omicron variants exhibited some changes in primers and probes. This nucleotide change pattern was corroborated with a couple of stem-loops formed in secondary structure that was the same to all variants. Notably, mutations in the N1 region of these three variants were located in the first stem-loop or spacer region, potentially leading to false-negative results due to their proximity to the hybridization region of primers and probes used in RT-qPCR, but without affecting their structure.
Moreover, in silico results with a 100% frequency threshold indicated that only the N2 region from Gamma, GH490R, and Omicron variants could be correctly identified, whereas the other variants and the Nl region from all variants had a high probability of generating false-negative results 35, particularly the Delta variant as reported previously 36.
The above was supported by the individual RT-qPCR results from 20 saliva samples, where two samples did not show the expected result for the N1 region but tested positive for the N2 region. Therefore, the N1 region of both samples could correspond to any nucleotide combination from SARS-CoV-2 variants without 100% identity with both frequency thresholds. In a RT-qPCR experiment, two reactions may show similar Ct values but different fluorescence signals, under different conditions37. Moreover, the implementation of 60 mM of TMAC, improves Illumina sequencing libraries preparation from AT-biased genomes38 and avoids non-specific amplification in the Loop-mediated isothermal amplification, LAMP39. In addition to above, it has been demonstrated that the addition of 3-5% of DMSO in PCR reactions, enhances the amplification of regions with high GC% content40,41.
In accordance with that, even though Ct values did not show statistically significant differences, which may be due to the viral load of the sample or the integrity of the viral RNA and the presence of canonical and non-canonical sub-genomes42; an increase in fluorescence signal was observed when DS was implemented, which indicates that the DS improves the polymerization signal despite the RNA amount present in the sample. To confirm this, sequencing of the samples is necessary to establish the nucleotide organization and potential variant presence.
These findings were further validated with multiplex RT-qPCR in pooled samples, where the fluorescent signal was proportional to the increase in positive samples compared to negatives. This result was attributed to the fact that the probes for the N1 and N2 regions had the same fluorophore [FAM] in the same RT-qPCR, leading to a cumulative increase in signal with each RT-qPCR cycle.
Considering that SARS-CoV-2 is primarily transmitted through saliva aerosols generated during coughing, sneezing, and speaking 17,43, obtaining saliva samples for detection through RT-qPCR provides a representative and cost-effective solution with a substantially increased patient acceptance compared to nasopharyngeal sampling, as demonstrated previously 17,18,20.
Conclusion
The addition of DS in RT-qPCR for individual or pooled saliva pooled samples offers a rapid, simple, and easily applicable improvement in polymerization signal, thereby contributing significantly to the SARS-CoV-2 CDC procedure by eliminating RNA stem-loop structures from the N gene regions employed for its detection.

