











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
1. Introducción
Colombia alberga una gran diversidad de insectos vectores hospedadores, lo cual representa una oportunidad para investigar la propagación de enfermedades infecciosas y desarrollar medidas preventivas. Este escenario permite abordar la ecología y la epidemiología de las enfermedades transmitidas por vectores, para desarrollar estrategias de control tanto en Colombia como en otros países. Además, la caracterización de esta diversidad es un paso crucial para impulsar la investigación sobre nuevos tratamientos y vacunas que puedan aliviar la carga de enfermedades transmitidas por vectores [1].
La reproducción de los insectos puede verse favorecida por la predominancia de zonas geográficas de clima cálido en Colombia que tienen una temperatura media de 28°C sin grandes variaciones estacionales anuales. Estas zonas son apropiadas para el desarrollo y reproducción de los vectores de enfermedades tropicales [2] de dos taxones de importancia en salud pública, hemípteros y dípteros. Entre algunos miembros de estos grupos de importancia médica destacan Anopheles sp (malaria), Aedes sp (dengue, chikunguña y zika) y Triatoma sp (chagas) [3], entre otros.
Los insectos vectores, como vehículos de transmisión de enfermedades a humanos y a otros animales [4], se distribuyen en todas partes del mundo, por lo tanto, su biogeografía es un campo importante de estudio [5, 6]. La distribución de estos organismos, permiten comprender las interacciones intra e inter especies en su entorno, y así como el efecto de cambios físicos como el clima o el cambio de uso de suelo, sobre ellos [7]. En este sentido, estudiar la biogeografía de los insectos vectores-hospederos, permite diseñar mejores medidas de salud pública para mitigar la dispersión de enfermedades.
Otra manera de estudiar la diversidad de los insectos vectores-hospederos, es a través de la filogenia, acercamiento importante a considerar cuando se estudia la propagación de enfermedades. Al comprender las relaciones entre las diferentes especies de insectos y sus huéspedes, se esclarecen los mecanismos de transmisión de las enfermedades, y se desarrollan estrategias para prevenirlas [8]. Además, de plantear hipótesis que identifiquen nuevos vectores potenciales, eficientes en la transmisión de enfermedades [9]. Sin embargo, en Colombia hay un gran vacío de conocimiento en el área, la cual parece estar restringida a estudios basados sobre la taxonomía clásica. Por lo tanto, es pertinente evaluar la filogenia de los insectos vectores-hospederos a partir de marcadores moleculares comunes utilizados en publicaciones de acceso libre, así como la distribución geográfica potencial de estos insectos en el territorio nacional.
La clasificación taxonómica eficiente y rápida de estos vectores es importante en la caracterización epidemiológica de las enfermedades infecciosas que transmiten. Por esta razón, se ha avanzado en la caracterización molecular a través de marcadores moleculares, de estos insectos, como un método complementario a la taxonomía clásica [10]. En el caso de la presente investigación, se utilizará el concepto para referirse a una secuencia de ADN (genes o partes de ellos) [11], utilizados para determinar la variación genética de una población de organismos en el tiempo. Los marcadores moleculares son la caja de herramientas más utilizada en la caracterización e identificación de los seres vivos, y su uso es cada vez más común en la identificación, clasificación y evolución de los insectos, así como de sus simbiontes o parásitos acompañantes [12]. De este modo, los científicos y los funcionarios de salud pública pueden comprender los mecanismos de propagación de enfermedades transmitidas por vectores para desarrollar estrategias más eficaces en su control epidemiológico.
La caracterización molecular se puede tomar como rasgos ge-notípicos que complementan los patrones taxonómicos para la clasificación de los organismos vivos. Entre los marcadores moleculares más comunes en estudios de eucariontes están COI (citocromo C oxidasa subunidad I), COII (citocromo C oxidasa subunidad II), ITSs (espaciador transcrito interno) 1 y 2. Los genes COI y COII (Fig. 1 A) codifican para dos de las siete subunidades polipeptídicas del complejo citocromo c oxidasa, quien hace parte la cadena transportadora de electrones en las mitocondrias de organismos eucarióticos [13]. Mientras que, la región espaciadora interna transcrita del ADN ribosomal (ITS) ha sido útil para estudios de relaciones filogenéticas por su alta tasa de sustitución. La región de los ITS en eucariontes comprende el ITS2, y el ITS1 (Fig. 1B). Donde el ITS1 es el separador de la subunidad 18S y la subunidad 5.8S, mientras que el ITS2 es el separador entre la subunidad 5.8S y subunidad la 28S [14]. Sin embargo, en la caracterización molecular también se han utilizado otros marcadores como el gen de la esterasa, gen de la acetilcolinesterasa, gen de la NADH deshidrogenasa, y el gen de la apolipoproteína [15]. Estos genes junto con COI, muestran poca tasa de sustitución por su naturaleza codificante, rasgo que no permite una clara resolución taxonómica si se utilizan individualmente.
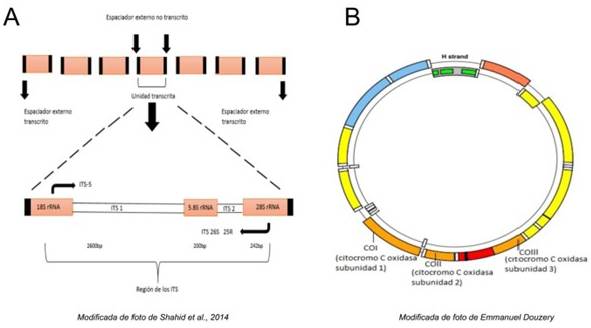
Figura 1: (A) Se muestra en detalles los espaciadores internos de transcritos (ITS). Figura modificada de Shahid et al., 2014 [16]. (B) ADN mitocondrial mostrando las regiones del gen GEN CITOCROMO OXIDASA. Figura modificada de Emmanuel Douzery en Wikipedia.
Con el objetivo de evaluar la adecuación de un marcador molecular específico en relación con la clasificación taxonómica clásica, en este estudio se llevó a cabo la construcción de árboles filogenéticos utilizando el método de Máxima Verosimilitud. Se utilizaron secuencias publicadas de los marcadores más comúnmente empleados en la caracterización molecular de insectos, como COII e ITS2, que son de particular interés en los órdenes Hemiptera y Diptera, los cuales tienen una relevancia médica y epidemiológica significativa. Para llevar a cabo estos análisis, se utilizó el software Mega X[17].
Los hallazgos obtenidos en este estudio revelan la identificación filogenética y la distribución geográfica de insectos vectores clave, tales como Aedes aegypti, Anopheles sp, Rhodnius prolixus y Lutzomia sp. Estos insectos son responsables de transmitir enfermedades de importancia médica, como el dengue, la fiebre amarilla, la malaria, la enfermedad de Chagas y la leishmaniasis, entre otras. La información obtenida, junto con su distribución geográfica, adquiere gran relevancia debido a que el enriquecimiento de las bases de datos moleculares de estos insectos vectores permitirá realizar aproximaciones taxonómicas, evolutivas y funcionales más precisas a lo largo del tiempo y el espacio. Esto, a su vez, contribuirá a desarrollar estrategias de control más efectivas y precisas para abordar el problema de salud pública que representan los insectos vectores en Colombia, así como a nivel mundial.
2. Métodos
Distribución potencial de Diptera y Hemiptera en Colombia
Para las búsquedas de las coordenadas geográficas, se realizó mediante los registros que había en el SiB Colombia SiB Colombia (2020, abril 10). Se filtró la búsqueda por especies, iniciando con el orden Hemiptera para una sola búsqueda, y después con el orden Díptera. Esta búsqueda arrojó una base de datos GBIF.org (2020), luego se descargó en formato.mtsx (material suplementario 14/15/16/17: ITS2 Hemiptera/ITS2 Diptera/COII Hemiptera/COII Diptera.mtsx), la cual se utilizó para la construcción de un mapa de distribución potencial para estos insectos, por medio del programa ArcGIS versión 10.5 [18]. La ubicación de los insectos registrados con información molecular trabajados en esta investigación, se agregaron manualmente en el área especificada en la información de cada una de las secuencias encontradas (material suplementario: Base de Datos Insectos Portadores de Vectores.xlsx).
Construcción de base de datos local, y relaciones filogenéticas
La selección de las secuencias de los genes se realizó teniendo en cuenta los marcadores moleculares como herramienta para estudios filogenéticos, COII e ITS2. Se realizó una búsqueda de los marcadores anteriormente nombrados en la base de datos NCBI (Centro Nacional para la Información Biotecnológica), para especies del orden Diptera y Hemiptera reportados para Colombia, excluyendo secuencias de otros países (material suplementario 1: Base de Datos Insectos Portadores de Vectores.xlsx).
Reconstrucción filogenética
Las secuencias obtenidas se descargaron en formato FASTA (material suplementario 2/3/4/5: Hemiptera ITS2/Diptera ITS2/Hemiptera COII/Diptera COII.FASTA), se alinearon en el programa MEGA mediante el algoritmo Muscle (Tamu-ra et al., 2013). Después, se exportaron en formato. Meg (material suplementario 6/7/8/9: Hemiptera ITS2/Diptera ITS2/Hemiptera COII/Diptera COII.Meg) para la construcción de la filogenia molecular por medio de los modelos estadísticos de probabilidades de máximum likelihood y Neighbor-Joining, utilizando para cada árbol el modelo evolutivo que mejor se ajustara a los datos [19]. Se utilizaron 1000 repeticiones Bootstrap, teniendo en cuenta transiciones y transversiones para cada árbol, y para cada marcador molecular.
Se reconstruyeron los árboles con los mismos marcadores, utilizando el método de máxima verosimilitud, el cual evalúa todos los posibles árboles que representen la evolución y busca el más óptimo, posteriormente se calculó la distancia genética mediante una matriz generada por el mismo software (MEGA X). Se utilizaronaproximadamente 3000 secuencias con los diferentes marcadores moleculares ITS2, y COII encontradas en la base de datos NCBI en el Genbank (material suplementario 10/11/12/13: Models G Hemiptera ITS2/Models G Diptera ITS2/Models G Hemiptera COII/Models G Diptera COII.xlsx). Además, se eliminaron todas las posiciones con menos del 95 % de cobertura del sitio, es decir, se permitieron menos del 5 % de brechas de alineación, datos faltantes y bases ambiguas en cualquier posición (opción de eliminación parcial).
3. Resultados y discusión
Distribución potencial de Diptera y Hemiptera en Colombia.
La integración de la densidad de registros que refleja la distribución potencial de los insectos vectores-hospederos con secuencias de marcadores moleculares en el Genbank, para Colombia se observa en la figura 2.
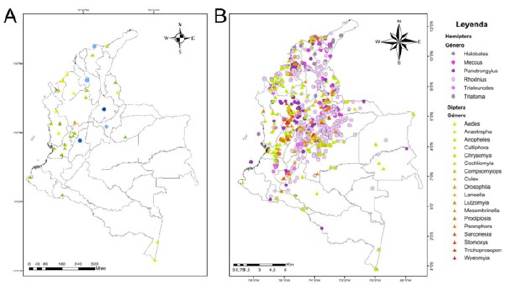
Figura 2: Distribución geográfica de los lugares donde se han realizado estudios A. Distribución geográfica de los vectores insectos-hospederos con los marcadores moleculares COII e ITS2 en Colombia, registros extraídos de la base de datos NCBI. B. Distribución geográfica obtenida de la base de datos SiB.
En consecuencia, Diptera está presente en diferentes partes de Colombia, como Turbo-Antioquia, Chinchina-Caldas, Charambina Bosque-Choco, Medio San Juan-Chocó, Riofrío-Valle del Cauca, Pereira-Risaralda, Nechi-Antioquia, Valle del Cauca-Buenaventura, Córdoba-Puerto Libertador, Tibú-Norte de Santander, Arboletes-Antioquia, Dibulla-Guajira, Necocli-Antioquia, Chocó-Nuqui, El Bagre-Antioquia, Tara-paca -Amazonas, Leticia-Amazonas, Monitos-Córdoba, San Pedro de Urabá-Antioquia, Zulia-Norte de Santander, Los Achiotes-Magdalena, Dibulla-La Guajira, Cartago-Valle del Cauca, Chocó, Guarumito-Casanare y Puerto Asís-La Manuela (Fig. 2 A). Mientras que Hemiptera está presente en Boyacá, Sucre, Sierra Nevada- Santa Marta y Tolima (Fig. 2 A).
Cabe anotar que los registros con información molecular (Fig. 2 A) muestran una cantidad menor en comparación con los registros basados en taxonomía clásica (Fig. 2 B). Esto sugiere la existencia de una escasez de datos moleculares en el territorio nacional. Además, seres alta que el género Rhodnius del orden Hemiptera es el más representativo en la región andina, mientras que se encuentra en menor concentración en la Amazonia. En este sentido, es importante considerar que la distribución de Diptera y Hemiptera en el noroeste del país puede estar influenciada por un mayor esfuerzo de muestreo en esta zona y no necesariamente reflejar una distribución real.
En conjunto, los datos de distribución potencial de los órdenes Hemiptera y Diptera en Colombia indican una carencia de información molecular de las especies de estos órdenes en el país, así como posibles deficiencias en la georreferenciación de los muestreos que proporcionan los datos moleculares. Además, se observa una falta de registros basados en taxonomía clásica, también georreferenciados, de las especies que componen estos órdenes en la región sureste del país. Estos hallazgos resaltan la necesidad de unir esfuerzos para realizar investigaciones enfocadas en conocer la diversidad y la distribución de especies de insectos vectores-hospederos en toda la geografía colombiana, así como para establecer una base nacional de biodiversidad donde se puedan depositar estos datos.
Relación filogenética de Diptera
Aunque el grupo de datos utilizados en este estudio se extrajo de diferentes fuentes bibliográficas en las cuales utilizaban un marcador molecular individual según sus necesidades, se logró hacer comparaciones filogenéticas con resultados interesantes (Fig. 3).
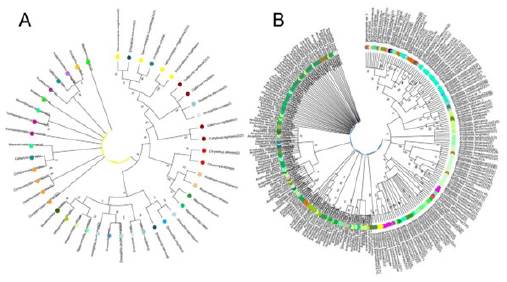
Figura 3: Árboles filogenéticos de máxima verosimilitud y el modelo Tamura-Nei para Diptera. A. Árbol filogenético utilizando el marcador COII. Se muestra el árbol con la probabilidad logarítmica más alta (-4499,26). Se utilizó una distribución Gamma discreta para modelar las diferencias de tasas evolutivas entre sitios (2 categorías (+G, parámetro = 0,2789)). Este análisis involucró 45 secuencias de nucleótidos. y hubo un total de 589 posiciones en el conjunto de datos final. B. Árbol filogenético utilizando el marcador ITS2. Se muestra el árbol con la probabilidad logarítmica más alta (-17936,62). Este análisis involucró 259 secuencias de nucleótidos y hubo un total de 949 posiciones en el conjunto de datos final. El porcentaje de árboles en los que los taxones asociados se agruparon se muestra junto a las ramas. Los árboles iniciales para la búsqueda heurística se obtuvieron automáticamente aplicando los algoritmos Neighbor-Join y BioNJ a una matriz de distancias por pares estimadas mediante el modelo Tamura-Nei y luego seleccionando la topología con un valor logarítmico de verosimilitud superior. Al final de cada rama se encuentra el respectivo nombre de cada especie acompañado de un número entre paréntesis indicando el total de veces presentes de la misma secuencia en la base de datos construida de la información tomada en el GenBank®. Los análisis evolutivos se realizaron en MEGA X.
La construcción de los árboles filogenéticos con los marcadores moleculares COII e ITS2 muestran lo siguiente: El árbol de máxima verosimilitud para el orden Diptera con el marcador COII consta de 18 clados parafiléticos con una resolución mayor al 55 %. Es de resaltar, la agrupación de diferentes géneros en un mismo clado con un porcentaje alto de soporte. Por ejemplo, Sarconesiopsis y Drosophila nebulasa, a pesar de ser dos géneros diferentes, se agruparon en el mismo clado con un soporte de 99 %. Por otra parte, al utilizar el marcador molecular ITS2 para la construcción del árbol filogenético para el mismo orden, se puede observar que el orden Diptera se compone de 28 clados parafiléticos con soportes por encima del 50 %. Este análisis filogenético, también agrupa diferentes géneros en el mismo clado, por ejemplo, Anopheles y Culex, hacen parte del mismo clado con un soporte del 75 %.
Los resultados de los árboles de máxima verosimilitud permiten inferir que las relaciones filogenéticas a nivel interpo-blacional de las especies de Diptera analizadas aquí, tienen un origen monofilético, reafirmando así la clasificación taxonómica clásica actual [17]. En el cladograma obtenido con el marcador molecular COII de Diptera (Fig. 3A), se aprecia una politomía en la mitad basal del árbol, esto puede deberse a la falta de datos para establecer una clara relación entre estos grupos o puede ser evidencia de una rápida evolución genética de éstos.
La filogenia a nivel de género muestra aparentemente no ser monofilético. Sin embargo, este resultado se debe a un error en sí del modelo al "interpretar" convergencias como sinapomorfías [20]. La mayoría de las especies de Drosophila forman un grupo junto a algunas especies de Mesembrinella, el cual, es un clado con fuerte soporte (99 %). Otras especies de Drosophila forman clados con especies de Calliphora y Sarconesiopsis. Especies de Chrysomira, Roraimusca y especies de Mesembrinella forman un clado con un alto soporte (96 %). La presencia de una gran politomía en este árbol, muestra que el marcador COII no es el mejor proxy para establecer relaciones filogenéticas claras dentro de los géneros de Diptera. Por lo tanto, se deben utilizar más marcadores mitocondriales para asemejar la tasa de sustitución de un marcador nuclear como el ITS. Sin embargo, si se utiliza el marcador ITS2 es necesario acompañar los resultados con análisis de taxonomía clásica y ecología de la especie o género estudiado.
En el cladograma construido con el marcador molecular ITS2 de Diptera (Fig. 3B), también se aprecia una politomía con numerosas especies de Anopheles y algunas especies de Culex. El género mejor representado en el cladograma fue Anopheles, este género forma clados con otros géneros, con diferentes valores de soporte. La presencia de múltiples politomías puede ser evidencia de un origen reciente de las especies de estos géneros producto de una rápida radiación [21]. Además, se evidenció que Cochiliomya y Prodiplosis son grupos hermanos con respecto a Anopheles. Mientras que Mesembrinella es un grupo hermano de Culex con respecto a Huascaromusca.
Relación filogenética de Hemiptera
En cuanto a los árboles filogenéticos de máxima verosimilitud para el orden Hemiptera usando el marcador COII, se evidencian 20 clados parafiléticos con una resolución mayor al 13 %, agrupando diferentes géneros en un solo clado, como se logra demostrar en el caso de Ventidius y Esakia, que muestran una resolución del 50 % (Fig. 4). En cuanto al marcador ITS2, exhibe 14 clados parafiléticos con una resolución mayor al 50 %, agrupando así diferentes géneros en un solo clado, como el caso de los géneros Pastrongylus y Rhodnius, con una resolución del 100 % (Fig. 4).
El árbol filogenético de Hemiptera construido con el marcador molecular COII (Fig. 4A), como en el caso de Diptera, exhibe múltiples politomías. Lo anterior, no permite establecer relaciones claras dentro de las especies pertenecientes a los géneros de este orden [22]. Los géneros Metrocoris, Eurymetra, Acutaspis, Asclepios son monofiléticos, con suportes entre 63 % y 99 %. Metrocoris, y Eurymetra forman una politomía junto a Ptilomera, Ventidius, Halovelia y Esakia. El género mejor representado en el cladograma fue Halobates, pero su relación con otros géneros no queda bien establecida, evidencia de ello son las múltiples politomías, siendo necesario más estudios para establecer bien estas relaciones. En cuanto al árbol filogenético construido con el marcador molecular ITS2 de Hemiptera (Fig. 4B), muestra que Triatoma forma una gran politomía, tal vez, por la sobrerrepresenta-ción en secuencias de Traitoma dimidiata. El género Triatoma parece ser parafilético, pero forman un clado monofilético con la inclusión de especies de los géneros Rhodnius y Panstrongylus, con un soporte en el límite de confianza (50 %). Los géneros Rhodnius y Panstrongylus muestran ser polifiléticos con múltiples especies anidadas dentro del género Triatoma. Estos resultados son congruentes con los obtenidos por otros autores, en donde explican que estas agrupaciones podrían ser artefactos debido a problemas en el modelo, al tomar convergencias como sinapomorfías [1].
A pesar de que la región ITS2 es un biomarcador genético informativo para establecer y demostrar las relaciones filogenéticas de un género o familia, se recomienda la inclusión de otros genes o el genoma completo para realizar inferencias filogenéticas confiables que permitan esclarecer las relaciones evolutivas que pueden presentarse en las familias de los insectos estudiados aquí [23]. El marcador ITS2 se ha utilizado en la identificación de miembros en los órdenes Diptera y Hemiptera [24], pero en Colombia se posee escasa información para realizar estudios comparativos. Además, las especies tanto para hemípteros y dípteros con información molecular reportada y estudiadas en esta investigación, solo cuentan con la información de uno u otro marcador molecular, lo cual dificulta realizar una comparación precisa entre la resolución filogenética que pueda dar el marcador molecular COII o ITS2, ya que los grupos de insectos caracterizados por ITS2 no son los mismos caracterizados por COII y viceversa. Sin embargo, esta situación es una gran oportunidad para potenciar este tipo de investigaciones en el país.
Alcances y limitaciones de los marcadores.
Los marcadores moleculares COII e ITS2 históricamente han sido la herramienta predilecta para estudios de filogenia y evolución [25]. Sin embargo, existen otros que pueden ser útiles para complementar estos enfoques en los taxones de insectos estudiados aquí [22]. Genes nucleares como los que codifican para la esterasa, la acetilcolinesterasa y apolipo-proteína, y el gen mitocondrial que codifica para la NADH deshidrogenasa, se han utilizado como marcadores para realizar acercamientos evolutivos entre diferentes especies, así como para analizar la variación genética dentro de especies de insectos hospederos [26]. En este estudio no se tomaron en cuenta estos marcadores moleculares, debido a que solo se quería comparar las hipótesis que se podían construir con un marcador molecular estructural como el ITS2, el cual está sujeto a mayores tasas de sustitución que cualquier gen codificante como los nombrados anteriormente, así como el uso de la contraparte hereditaria materna, representado por el marcador molecular mitocondrial clásico COI II. En este sentido, es importante resaltar que las filogenias son hipótesis de las relaciones evolutivas de los taxones que se investigan [27]. Por lo tanto, en este trabajo se representan relaciones filogenéticas probables construidas con un método estadístico robusto, de los géneros Hemiptera y Diptera, en donde se muestra que los árboles filogenéticos más a fin a la clasificación taxonómica clásica, fueron los construidos con la región intergénica de los genes estructurales ribosomales.
Por otro lado, los mapas de distribución potencial de los géneros de insectos trabajados en este estudio, fueron construidos con los datos disponibles en SiB Colombia 2022, filtrando la búsqueda y obteniéndose la base de datos del GBIF (Infraestructura Mundial de Información en Biodiversidad) de colectas y clasificaciones taxonómicas clásicas, y de los datos sustraídos de la información depositada en cada una de las secuencias ITS y COII utilizadas en las construcciones filogenéticas realizadas aquí. Por tal razón, se podría estar dando un resultado sesgado y disminuido, debido a que no se incluyen datos actualizados. Sin embargo, muestra la pertinencia de realizar estos censos con la debida recolección de puntos GPS y descripciones geográficas de colecta.
4. Conclusiones
Las relaciones filogenéticas de los vectores-hospederos entre las secuencias obtenidas de algunas especies que componen tanto al orden Diptera como al Hemiptera, permiten inferir que estos insectos tienen un origen monofilético a nivel de género, y en algunos casos, a nivel de familia. Sin embargo, se formaron algunos clados que incluían especies de diferentes géneros, mostrando una baja resolución filogenética a este nivel taxonómico, lo cual puede ser explicado por una interpretación errónea de convergencias como sinapomorfías. Además, se muestra que el marcador molecular ITS2, da una mejor resolución de los géneros en clados individuales, tanto en el orden Diptera como en Hemiptera reportados hasta el momento en Colombia. Sin embargo, esta aseveración se debe tomar con cuidado, ya que los insectos estudiados con el marcador ITS2 no fueron los mismos que los reportados con COII, lo cual hace evidente la necesidad de realizar estudios en los cuales se incluyan mínimo a estos dos marcadores junto con otros marcadores mitocondriales, o al genoma completo. Por lo tanto, se recomienda complementar estos estudios con un número mayor de marcadores moleculares, con genomas completos, y/o específicos para cada género de interés, de la mano de estudios taxonómicos clásicos y ecológicos, con el fin obtener resultados más precisos y coherentes. Los marcadores son herramientas importantes para comprender la dinámica de las enfermedades transmitidas por vectores en esta región, ya que proporcionan una forma de rastrear cómo las diferentes especies de vectores interactúan entre sí. Al estudiar la diversidad genética de vectores en Colombia, se puede comprender mejor cómo se propagan estas enfermedades y desarrollar estrategias para controlarlas.
Los marcadores, ITS2 y COII son los más utilizados en Colombia para la identificación de organismos. Esto se debe principalmente a su alta estabilidad molecular, su facilidad de amplificación por PCR, su capacidad para detectar variaciones genéticas entre diferentes poblaciones y su relativa sencillez para ser obtenidos. Además, estos dos marcadores son los más adecuados para la identificación de especies en Colombia ya que otros marcadores como los mencionados anteriormente no son tan precisos como los utilizados. Por otro lado, se evidencia que los insectos estudiados en este trabajo, se distribuyen por gran parte del territorio nacional, sobre todo en el centro y norte del país. Sin embargo, los datos moleculares son escasos y los registros taxonómicos son bajos en el sureste del país. Con lo anterior se requiere unir esfuerzos para estudiar la biodiversidad tanto molecular como taxonómica de insectos vectores-hospederos, y crear una base de datos nacional de libre acceso.
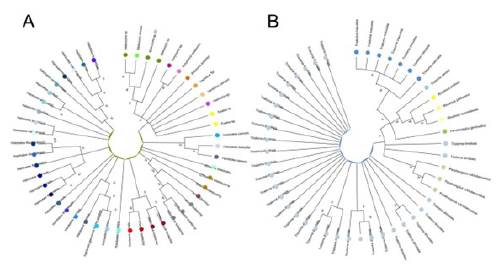
Figura 4: Árboles filogenéticos de máxima verosimilitud y el modelo Tamura-Nei, para Hemíptera. A. Árbol filogenético de máxima verosimilitud, marcador COII. Se muestra el árbol con la probabilidad logarítmica más alta (-19660,40). El porcentaje de árboles en los que los taxones asociados se agruparon se muestra junto a las ramas. Los árboles iniciales para la búsqueda heurística se obtuvieron automáticamente aplicando los algoritmos Neighbor-Join y BioNJ a una matriz de distancias por pares estimadas mediante el modelo Tamura-Nei y luego seleccionando la topología con un valor logarítmico de verosimilitud superior. Este análisis involucró 49 secuencias de nucleótidos. Hubo un total de 1439 posiciones en el conjunto de datos final. B. Árbol filogenético utilizando el marcador ITS2. Se muestra el árbol con la probabilidad logarítmica más alta (-4612,15).. Este análisis involucró 47 secuencias de nucleótidos. Hubo un total de 1282 posiciones en el conjunto de datos final. El porcentaje de árboles en los que los taxones asociados se agruparon se muestra junto a las ramas. Los árboles iniciales para la búsqueda heurística se obtuvieron automáticamente aplicando los algoritmos Neighbor-Join y BioNJ a una matriz de distancias por pares estimadas mediante el modelo Tamura-Nei y luego seleccionando la topología con un valor logarítmico de verosimilitud superior. Al final de cada rama se encuentra el respectivo nombre de cada especie acompañado de un número entre paréntesis indicando el total de veces presentes de la misma secuencia en la base de datos construida de la información tomada en el GenBank®. Los análisis evolutivos se realizaron en MEGA X.

