











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Temporal artery vasculitis in patients under the age of 50 years is an unusual form of vasculitis that manifests with temporal and ocular signs, with a group of etiologies that includes anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV),1 polyarteritis nodosa (PAN),2 hypereosinophilic syndrome (HES), amyloidosis,3 Kimura disease4 (KD), and angiolymphoid hyperplasia with eosinophilia (ALHE).5,6
Regarding the HES, the lymphoid-variant of hypereosinophilic syndrome (L-HES) presents aberrant helper T cells which produce interleukin (IL) 5, IL-4, and IL-13 that favor differentiation, activation, and eosinophilic infiltration. The LHES may have rheumatologic manifestations in up to 30% of cases, including variable vessel vasculitis.7 Herein, we present a case of left temporal arteritis caused by eosinophilic vasculitis (EV) associated with L-HES. This case report describes the clinical features, histology, and treatment of temporal arteritis in young patients and hypereosinophilic syndrome, as well as clues for their differential diagnosis.
Case report
A 36-year-old woman, without relevant past medical history or family health history, was evaluated in November 2021 because of hypereosinophilia (>1500 cells/|xl) of three years of evolution, associated with episodic facial swelling and pruritic hive-like lesions with residual post-inflammatory hyper-pigmentation located on the neck, supraclavicular region, shoulders, armpits, arms, and chest (Fig. 1). In addition, the patient reported arthralgias in elbows, wrists, knees, and ankles, with no infectious etiologies identified.
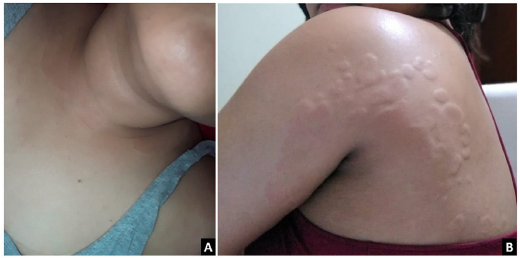
Fig. 1 (A) Erythematous plaques on the supraclavicular region and the left shoulder. (B) Multiple hives of different sizes converge toward the left axillary region.
Initially, her condition was considered an allergic reaction with elevated immunoglobulin E (IgE). She received multiple antihistamines, montelukast, and short cycles of oral steroids (methylprednisolone 20 mg/day), achieving temporary skin involvement improvement but persistent eosinophilia. Hence, in 2019 a bone marrow aspiration (BMA) with biopsy was performed, evidencing an increase in eosinophil precursors (7.6%) without dysplasia or other abnormalities. The flow cytometry in bone marrow showed non-clonal hypereosinophilia in a percentage of 9.9%, and the fluorescence in situ hybridization (FISH) for FIP1L1-PDGFRA rearrangement was negative.
At the beginning of 2021, the patient perceived a subtle thickening in the left temporal artery with no associated symptoms. However, three months before her presentation (August 2021), she manifested left temporal headache radiating toward the left maxillary region, local dysesthesias, photophobia, and ipsilateral mandibular claudication. An outpatient brain computed tomography (CT) was normal, and the Doppler ultrasound of temporal arteries showed positive "halo" and "compression" signs in the left common superficial temporal artery (Fig. 2). With the former result, the patient was admitted to the emergency department. On physical examination, the left temporal artery was thickened compared to the contralateral, pulses were present, no skin lesions on the scalp were evident, and her ocular fundus and visual acuity were normal. Fig. 3 presents the timeline of events, and the Table 1 summarizes relevant laboratory data.
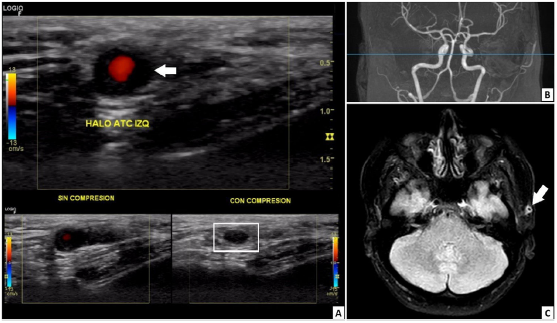
Fig. 2 (A) Doppler ultrasound image with a multifrequency linear transducer. Left common superficial temporal artery with hypoechoic, concentric, and homogeneous wall thickening well delineated toward the luminal side, up to 1.9 mm in the transverse plane, which corresponds to the halo sign (white arrow), and remains visible after extrinsic compression (white box below), configuring the non-compressible sign. No alterations were found in the right common superficial temporal artery, its branches, or axillary arteries. (B) 1.5-Tesla cerebral magnetic resonance angiography images did not detect vascular lesions. (C) Fluid attenuated inversion recovery (FLAIR) sequence in the axial plane. Homogenous wall thickening with homogeneous contrast uptake was observed in the left superficial temporal artery (white arrow), suggesting temporal arteritis.
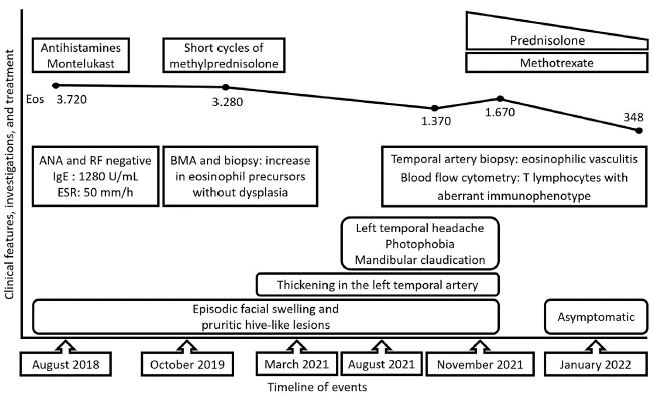
Fig. 3 Timeline of events. ANA, antinuclear antibodies; BMA, bone marrow aspiration; Eos, eosinophils per |xl; ESR, erythrocyte sedimentation rate; IgE, immunoglobulin E; RF, rheumatoid factor.
A cerebral magnetic resonance angiography (MRA) revealed vasculitis in the left superficial temporal artery without any other abnormal findings (Fig. 2). Chest CT, transthoracic echocardiogram, and thoracoabdominal MRA were normal. Additionally, a left superficial temporal artery biopsy evidenced panmural arteritis with marked eosinophilic infiltration (Fig. 4). Furthermore, a peripheral blood flow cytometry revealed a relative increase in eosinophils (7.1%) and 3.1% of T lymphocytes with aberrant immunophenotype (CD2+, CD3-, CD4+, CD5+++, CD45+), without other alterations. Moreover, a new BMA found a hypercellular bone marrow with increased eosinophil precursors (11.4%) without dysplasia. Bone marrow flow cytometry revealed T lymphocytes with the same aberrant immunophenotype detected in peripheral blood. The T-cell receptor rearrangement analysis could not be done because of insufficient material. Subsequently, left temporal arteritis caused by EV associated with L-HES was diagnosed.
The patient received 60 mg/day of prednisolone, achieving improvement of her symptoms. At discharge, methotrexate 15 mg/week was added. In the outpatient follow-up, she remained asymptomatic with improved eosinophilic count (Table 1), allowing the prednisone tapering.
Discussion
Temporal arteritis in patients under the age of 50 years is a rare form of vasculitis with different etiologies to the giant cell arteritis (GCA), the principal cause in patients above 50 years old but absent in younger patients.2 The "halo" and "compression" signs on Doppler ultrasound are usually associated with GCA but could be found in other vasculitis8 (Fig. 2).
Table 1 Laboratory data.
| Variable | 2018 | 2019 | On admission to hospitalization (November 2021) | At hospital discharge | Outpatient check-out (two months after hospitalization) | Reference range |
|---|---|---|---|---|---|---|
| White cells count (per µl) | 12,400 | 9,610 | 9,200 | 10,400 | 17,380 | 4,500-1,000 |
| Neutrophils (per µl l) | 4,092 | 2,840 | 4,040 | 4,760 | 9,559 | 1,500-9,000 |
| Lymphocytes (per µl l) | 2,860 | 2,940 | 3,080 | 3,390 | 5,909 | 1,500-4,000 |
| Monocytes (per µl l) | 420 | 410 | 620 | 540 | 1,043 | 30-900 |
| Eosinophils (per µl l) | 3,720 | 3,280 | 1,370 | 1,670 | 348 | 40-500 |
| Platelet count (per µl l) | 388,000 | 381,000 | 310,000 | 332,000 | 390,000 | 150,000-450,000 |
| Hemoglobin (g/dl) | 14.3 | 12.9 | 12.9 | 12.2 | 13.1 | 12-16 |
| ESR (mm/h) | 50 | 39 | 51 | |||
| CRP (mg/dl) | 1.1 | 1.0 | 0.4-1 | |||
| Scr (mg/dl) | 0.56 | 0.61 | 0.72-1.2 | |||
| IgE (U/ml) | 1,280 | 0-200 | ||||
| Vitamin B12 (pg/ml) | 292 | 211-911 | ||||
| LDH (U/L) | 508 | 391 | 120-246 | |||
| ANA by IFI | Negative | Negative | ||||
| ENA (U/ml) | Negative | <15 | ||||
| Anti-dsDNA by IFI | Negative | |||||
| RF (IU/ml) | Negative | <15 | ||||
| C3/C4 (mg/dl) | 101/24 | 116/35 | 90-160/10-50 | |||
| ANCA by IFI and ELISA | Negative | Negative |
ANA, antinuclear antibodies; ANCA, anti-neutrophil cytoplasmic antibodies; anti-dsDNA, anti-double stranded deoxyribonucleic acid antibodies; C3/C4, complement C3 and C4; CRP, C-reactive protein; ELISA, enzyme-linked immunosorbent assay; ENA, extractable nuclear antigen antibodies; ESR, erythrocyte sedimentation rate; IFI, indirect immunofluorescence; IgE, immunoglobulin E; LDH, lactate dehydrogenase; RF, rheumatoid factor; Scr, serum creatinine.
Among systemic vasculitis, eosinophilic granulomatosis with polyangiitis (EGPA) has been described as an etiology of temporal arteritis, and the onset at <50 years of age was found in five cases.1 On histological examination, EGPA presents significant infiltration by eosinophils without giant cells.1 However, our patient did not have a clinical presentation compatible with EGPA, and ANCA were negative. PAN has been described as a rare cause of temporal arteritis,9 but not in patients younger than 50 years of age. Light chain amyloidosis could infrequently compromise temporal arteries, mimicking giant cell arteritis.3 However, the absence of renal, cardiac, hepatic, or nervous system impairment was against this diagnosis in our case.
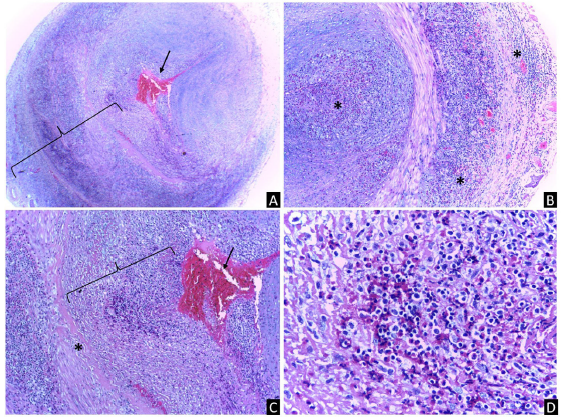
Fig. 4 (A) Left superficial temporal artery biopsy with arterial lumen obstructed by a fibrin thrombus (arrow) and inflammatory infiltrate, as well as a dense panmural inflammatory infiltrate (bracket) (original magnification 40 x). (B) Multifocal and massive eosinophilic inflammatory infiltrate (*) that involved the lumen, the entire vessel wall, and periadventitial tissues (original magnification 40 x). (C) Magnification from (A) with intraluminal fibrin thrombus (arrow), dense inflammatory infiltrate (bracket in the lumen), and fibrotic focus (*) in the intima (original magnification 100 x). (D) Magnification from (B) with profuse eosinophilic and lymphocytic infiltrate in the vascular wall (original magnification 400x ). The staining method was hematoxylin and eosin.
Other conditions such as KD and ALHE may resemble temporal arteritis (vasculitis mimickers) when they involve the temporal region.4,5 Both could manifest eosinophilia, elevated IgE, subcutaneous nodules or papules in the head and neck, and on histology, perivascular eosinophil infiltration with lymphoid follicular hyperplasia, in addition to conspicuous vascular proliferation.4,5 Nevertheless, in the described case, the clinical and histological features of KD and ALHE were absent, and these diseases do not explain the persistent hypereosinophilia.
On the other hand, HES is a heterogeneous group of disorders characterized by eosinophilic overproduction, leading to manifestations related to circulating cytokines, eosinophilic infiltration, and organ damage. Previous diagnosis criteria included eosinophil count >1500cells/µl for six months or more, eosinophil-mediated organ damage, and absence of other secondary etiologies.7 These criteria have raised criticism as most patients may require treatment before the onset of target organ damage or before six months.10
Current criteria for diagnosing HES include eosinophil blood count >1500 cells/ml on two occasions one month apart, presence of tissue or bone marrow eosinophilia, or marked deposition of eosinophil granule contents.7,11
Classically three major groups of HES can be distinguished:
Primary or clonal HES (HESn). The classification of eosinophilic diseases was revised in the 2008 World Health Organization (WHO) scheme of myeloid neoplasms and reaffirmed in 2016. It defined as primary causes of HES the category of "myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1 or with PCM1-JAK2", and from the category of myelo-proliferative neoplasms (MPNs) the "chronic eosinophilic leukemia, not otherwise specified" (CEL, NOS).7
Secondary or reactive HES (HESr), due to infections, allergy/atopy or hypersensitivity conditions, drug reactions, rheumatologic or pulmonary eosinophilic diseases, allergic gastroenteritis, metabolic conditions such as adrenal insufficiency, and non-myeloid malignancies (with the production of cytokines, such as IL-3, IL-5, and GM- CSF).7
Idiopathic HES, which requires exclusion of primary and secondary causes of hypereosinophilia.7
Specifically, L-HES is a HESr that presents an immunophenotypically aberrant T-cell population (CD3-CD4+ or double-negative T-cells CD3+CD4-CD8-), which produces interleukins (IL-5, IL-4, IL-13) that favor differentiation, activation, and tissue infiltration of eosinophils, without significant lymphocytosis. This entity may explain 17-27% of cases of unclassified hypereosinophilia in previous years and highlights immunophenotypes that may have rheumatologic manifestations, including vasculitis.1
Usually, patients with L-HES have cutaneous involvement, like pruritus, urticaria, and facial angioedema, as the primary manifestation,12,13 similar to this case report (Fig. 1). In a subgroup of patients with L-HES, cyclic episodes of angioedema and urticaria accompanied by increased serum IL-5 levels, IgM, and dramatic eosinophilia (known as Gleich's syndrome) have been described.12 Lefèvre et al.13 evidenced in 21 patients that L-HES could present multisystemic involvement, but it is usually heterogeneous, such as lymph nodes (62%), gastrointestinal (24%), pulmonary (19%), neurological (10%), and cardiovascular (5%) compromises.13 Rheumatologic involvement was identified in 29%, mainly carpal tunnel syndrome, polyarthralgia, and polyarthritis in hands, wrists, knees, ankles, and bilateral hand extensor tenosynovitis. However, this study did not report temporal artery involvement, like our patient.
HES could generate vascular involvement by wall invasion by eosinophils and release of cytotoxic proteins from their granules, responsible for inflammation (vasculitis), and subsequent necrosis of the vascular wall in vessels of any caliber.11 EV associated with HES has been defined as a separate entity distinct from EGPA.14 Two forms of EV have been described, both associated with the development of arterial and venous thrombosis. The first consists only of skin lesions such as pruritic papules, urticaria, and angioedema with mild eosinophilia. The second form presents cutaneous necrotizing eosinophilic vasculitis without features suggestive of EGPA.11 The differential diagnosis of HES and ANCA-negative EGPA can be challenging. Nonetheless, Leurs et al.15 performed a comparative study of 166 patients with EGPA or HES. They demonstrated that a low C-reactive protein level (<3.6 mg/dl) was a diagnostic biomarker suggestive of HES, like in the present clinical case (Table 1).
Lefèvre et al.16 conducted a retrospective study and literature review, identifying 117 asthma-free and ANCA-negative patients with HES and EV without specific HES etiology. The most common features were pruritic papular lesions, urticaria, purpuric papules, angioedema, or localized swelling. Hypereosinophilia was described in 69%, cutaneous EV in 34%, and thirteen cases (11%) presented temporal arteritis.16 Among the histopathological findings in the temporal artery biopsy, eosinophilic infiltrates were described in the medial layer, the intima, or in all three layers (panmural involvement), along with fibrin thrombus in the lumen,16 like in our case (Fig. 4).
Regarding thrombosis, thrombogenesis is promoted by hypereosinophilic conditions due to the capacity of the major basic protein (MBP) 1 to activate platelets with a potency comparable to thrombin17 and also due to direct damage to the endothelium, leading to thrombosis.11,14 Furthermore, active eosinophils maintain a high tissue factor expression,18 and the eosinophil cationic protein enhances factor XII activity and binding of endogenous heparinoids.11,17
To our knowledge, this is the first case of L-HES associated with vascular involvement only of the temporal artery. Previously, aneurysms of the coronary, cerebral and temporal arteries have been reported, and a temporal artery biopsy revealed a thrombotic aneurysm attributable to an epithelioid hemangioendothelioma. This lesion was associated with a perivascular eosinophilic infiltrate, without vascular wall infiltration.19
The goal of therapy in L-HES is to mitigate eosinophil-mediated organ damage, and glucocorticoids are the first-line therapy.7,20 Ogbogu et al.21 described the treatment of 188 patients with HES (17% had L-HES). The median dose of prednisone was 40 mg daily at onset, and most patients were maintained on corticosteroids (median dose of 10 mg) for two months to 20 years.21 Among 141 patients who received corticosteroid monotherapy, 85% had a response (complete or partial) after one month of treatment. Combination therapy with hydroxyurea and corticosteroids achieved response in 69% and 75% with interferon-α and corticosteroids.21 Strikingly, 11 patients received methotrexate, but 10 discontinued it due to lack of efficacy or toxicity.21 Likewise, in the study of Lefévre et al.,13 steroid-sparing management with methotrex-ate was initiated in 4 of the 21 patients, but none presented a response. Cyclosporine, tacrolimus, cyclophosphamide, and mepolizumab has also been used with good response.7,16,20 In our patient, the previous discontinued use of corticos-teroids could be the reason for the persistent eosinophilia that ended in a vasculitic complication. However, posterior treatment with methotrexate and glucocorticoids had a favorable response.
Conclusions
L-HES is an uncommon disease with clonal expansion of aberrant T lymphocytes producing an eosinophilic-poietic cytokine profile. The resultant hypereosinophilia could generate vasculitis phenomena, a rare form of presentation. The clinical features, imaging, and an adequate cytogenetic study allow differentiation from other forms of HES or systemic vasculitis. Although therapeutic options are limited, the goal is to mitigate eosinophil-mediated target organ damage by decreasing the eosinophil count and monitoring the risk of lymphoproliferative disorders. This report highlights the first case of L-HES with isolated involvement of a temporal artery and the rapid improvement with glucocorticoids and methotrexate.

