











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Interstitial lung diseases (ILDs) group more than 200 parenchymal pulmonary disorders, some of which have a progressive course with development of fibrosis, such as idiopathic pulmonary fibrosis (IPF) and hypersensitivity pneumonitis.1 Nevertheless, connective tissue disease-related ILD (CTD-ILD) is prevalent, representing around 30% of the non-IPF-ILDs. (2
ILD may be the first manifestation in autoimmune diseases or may appear during the disease. (3,4 A recent systematic review of 139 studies, addressed the prevalence of ILD in different autoimmune diseases, reporting 11% in rheumatoid arthritis (RA), 47% in systemic sclerosis (SSc), 41% in idiopathic inflammatory myopathies (IIM), 17% in Sjogren syndrome (Sjo) and 56% in mixed connective tissue disease (MCTD). (5
Regardless of the etiology of the non-IPF-ILD, some patients progress and develop fibrosis despite treatment, called as progressive pulmonary fibrosis (PPF) (6 characterized by progression of fibrosis on high-resolution computed tomography (HRCT), declining on pulmonary function tests, and worsening of symptoms. (7 The definition criteria for PPF8 is listed in Table 1.

RA and SSc are the autoimmune diseases most likely to have PPF. (1 Patients with PPF have a clinical presentation similar to IPF, with higher mortality rates than patients without progression. (8 For this reason, early identification of these cases is crucial, since antifibrotics may impact on the fibrosis progression. (9
In this systematic review, we aimed to identify the evidence available about epidemiology, risk factors, biomarkers and treatment options for PPF associated with autoimmunity.
Methodology
A systematic review of the literature was carried out. The search was limited to articles in English and Spanish. Original observational and descriptive articles, clinical trials, systematic reviews, and meta-analyses were included. Articles that provided incomplete information about patients with autoimmune diseases were excluded. After the systematic search, a manual search was carried out, reviewing the bibliographic references or related articles found during the searches in the different databases.
Based on the PRISMA (preferred reporting items for systematic reviews and meta-analyses) guidelines, the search was carried out in the following electronic scientific databases: PUBMED, EMBASE, SCOPUS, and LILACS, using the following MeSH terms as search terms: "Progressive fibrosing interstitial lung disease", "progressive fibrosing ILD", "PF-ILD" and keyword: "progressive fibrosing lung disease".
The results were filtered based on the title, a second filter by abstract, and a third filter by full text; this process was done in a double-blind and independent fashion by two researchers (JY and JC). After concluding this process, the results of each investigator were compared to identify similarities and differences. In case of discrepancies between the included studies of the first and second evaluators, a third investigator (DG) resolves the differences. Studies not retrieved in full text at the time of evaluation were excluded. Fig. 1 schematically presents the screening process developed and the number of references evaluated in each process activity.
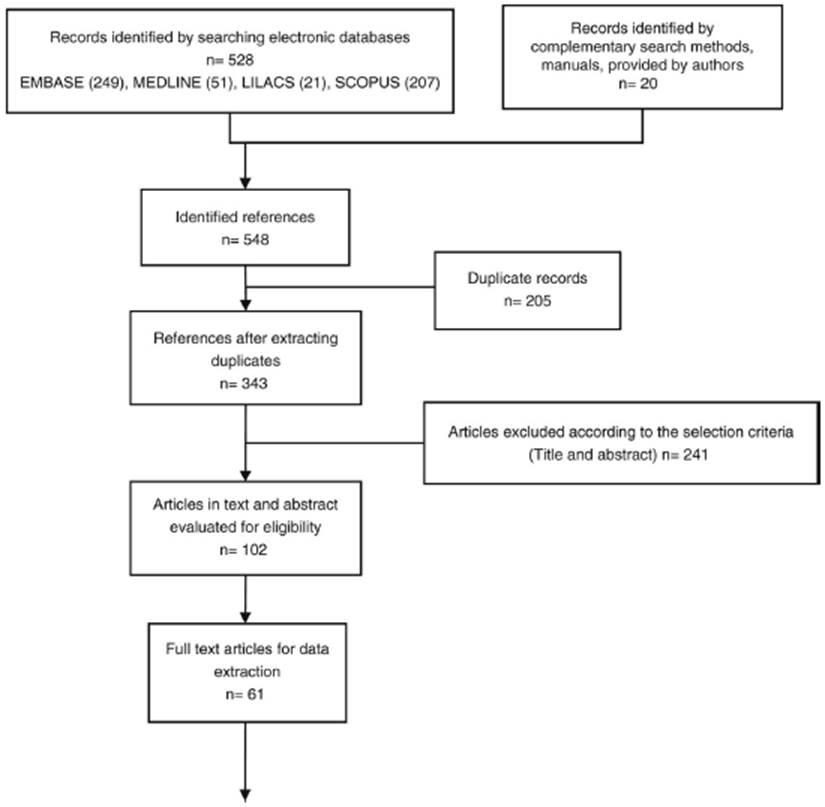
Source: adapted from: Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097, https://doi.org/10.1371/journal.pmed.
Fig. 1 PRISMA diagram.
In the screening and selection activity, 528 references were obtained after removing intrabase and interbase duplicates (n= 343). After the review by title and abstract, 102 references were obtained that were evaluated in the full text version, of which 61 original articles were used for data collection and extraction.
Results
There are few epidemiological studies about interstitial lung disease (ILD) and progressive pulmonary fibrosis (PPF) and even less data about CTD-ILD. Diagnosis of rheumatological diseases is not specified in most real-life studies and shows significant heterogeneity due to the variability of PPF definition, inclusion criteria, and recruitment of cases from diagnosis codes, records or prospective populations in real life.
The prevalence of ILD varies by country, it ranges from 6.3 to 76 per 100,000 people in Europe and 74.3 in the United States. (10 A recent study led by Hilberg11 in several European countries found a specific prevalence for ILD associated with rheumatoid arthritis (RA-ILD) of 0.4-3.1 per 100,000 inhabitants and 0.2-1.1 for other autoimmune diseases, as well as incidences of 1.1-18.1 and 1.1-15.9 per 100,000 inhabitants. This same study classifies each cause of ILD into subtypes. For RA-ILD, 11% had PPF, 27% had usual interstitial pneumonia (UIP) with a PPF, 33% UIP without PPF, and 29% had neither UIP nor PPF. For the other CTD-ILDs, the percentages were 15%, 25%, 9%, and 44%, respectively. In the whole population, including autoimmune or not autoimmune diseases, PPF, whether associated or not with UIP, represented 30%. Notably, sarcoidosis was the most prevalent diagnosis of ILD in this paper, in contrast to others, such as Hambly, (12 where it was only 4.5%, and CTD-ILDs were 44.5%. In additional reports, CTD-ILDs, irrespective of PPF have been reported in 67% of Kwon, (13 39.8% of Takei, (8 and 28.6%ofTorrisi. (14
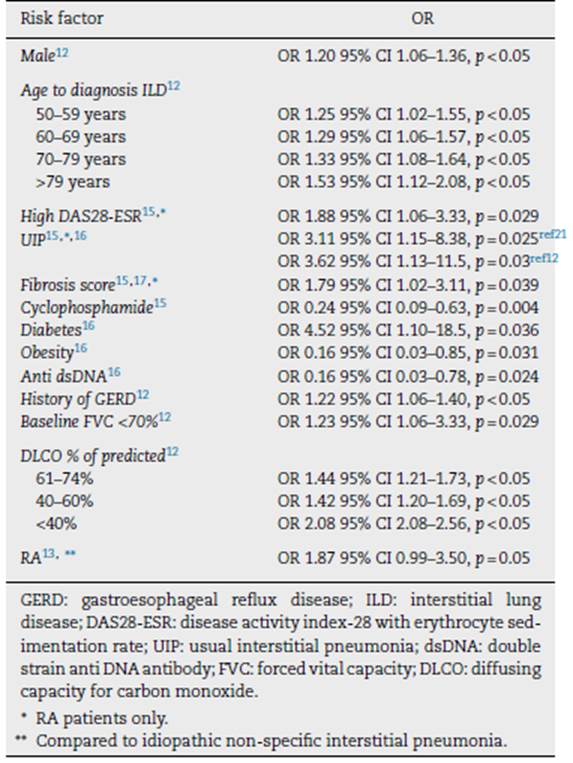
Identification of ILD progression is relevant due to its implications on mortality, therapy, and healthcare costs. Risk factors for progression to PPF in non-IPF-ILD are summarized in Table 2. Some of these studies only included patients with CTD-ILD, but others include different etiologies of fibrosing ILDs, such as hypersensitivity pneumonitis, sarcoidosis, pneumoconiosis, or non-classifiable ILDs.
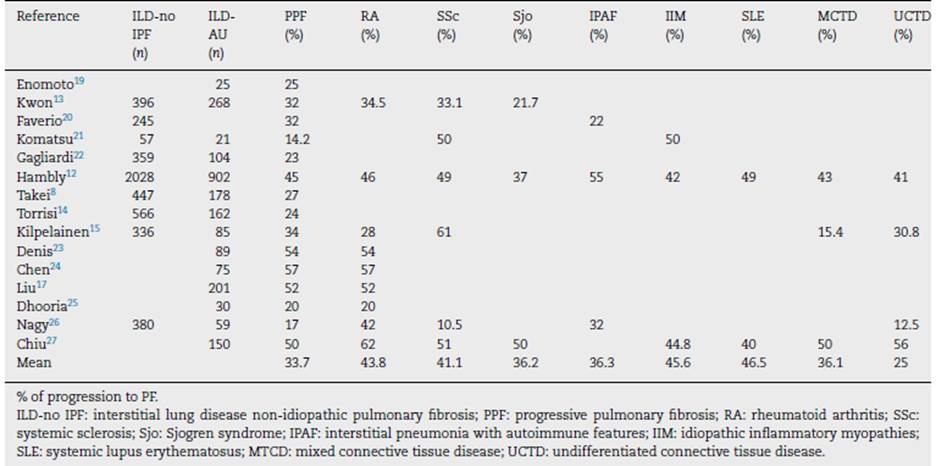
Most studies do not report the number of CTD-ILD patients who progress. Table 3 summarizes the data and the progression percentage for each type of autoimmunity to date. On average, the number of patients who progress is 33.7%, similar to what was found in the PROGRESS study, which does not separately provide data on autoimmune diseases. (18
Table 3 Progression of connective tissue disease-related interstitial lung disease (CTD-ILD) and each specific disease in different studies.
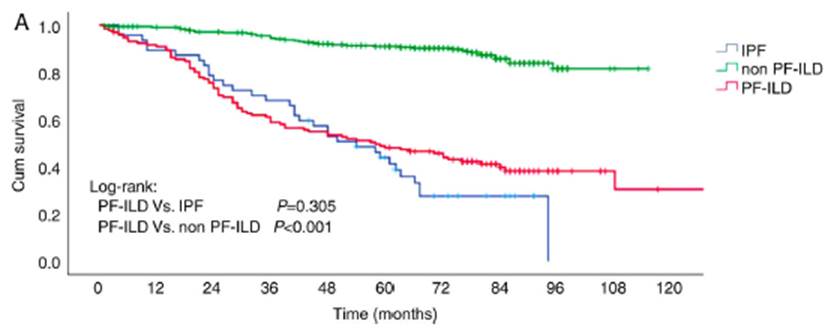
In the PROGRESS study, conducted in France, (18 the average survival in PPF non-IPF patients was 3.7 years; 95.2% required at least one hospitalization during the 7-year follow-up, and 34.3% required intensive care unit admission. Chen28 showed a survival rate at 1, 3, and 5 years of 90.9%, 58%, and 48%, respectively, which was comparable to the survival rate of IPF (log rank p = 0.30) but higher than the patients with non-IPF-ILD without PPF (log rank p < 0.001) (Fig. 2).
Faverio et al. (20 discovered similar differences in non-IPF-ILD without PPF, with a median survival time of 6 years and a mortality rate of 15% at 5 years, in comparison to patients with PPF who had a median 3-year survival time and mortality rates of 4 and 20% at 2 and 3 years, respectively.
Differences have been observed comparing the survival of non-fibrosing ILDs and fibrosing ILDs (p = 0.02) (21 or PPF (p = 0.001, (22p = 0.04, (21p = 0.00129). Similar survival rates between PPF to IPF have been reported in other studies (p = 0.51, (21p = 0.629) or even worse for PPF if IPF has no progression, according to Takei (p = 0.001). (8 The mortality risk of PPF-ILD across studies is summarized in Fig. 3.
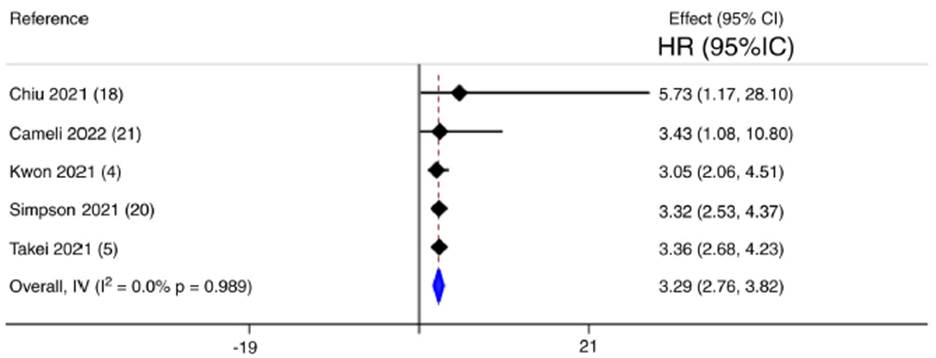
Table 4 summarizes the risk factors for death in the population of patients with autoimmune-related PPF-ILD.
Table 4 Mortality risk factors for patients with autoimmune progressive pulmonary fibrosis related interstitial lung disease (PPF-ILD).

On HRCT about 65% of people with systemic sclerosis (SSc) have parenchymal abnormalities, and about 40% will develop clinical ILD. ILD may cause death in 40% to 10% of SSc patients, making it the leading cause of death. (32 The most frequently identified pattern on HRCT is non-specific interstitial pneumonia (NSIP), followed by UIP. (33 Extension of the disease on HRCT and the low values of pulmonary function tests, as seen in Fig. 4, are correlated with higher mortality in comparison to patients with limited illness on HRCT (HR 3.46; 95% CI 2.19-5.46; p < 0.0005). (34
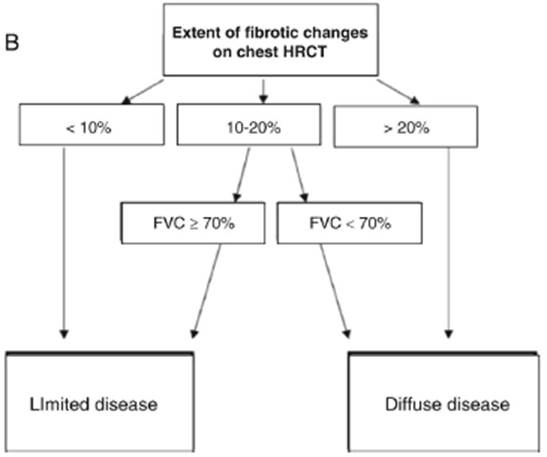
Source: modified from Goh et al. (34
Fig. 4 Definition of extensive and limited disease in SSc-ILD. HRCT: high-resolution axial tomography; FVC: forced vital capacity.
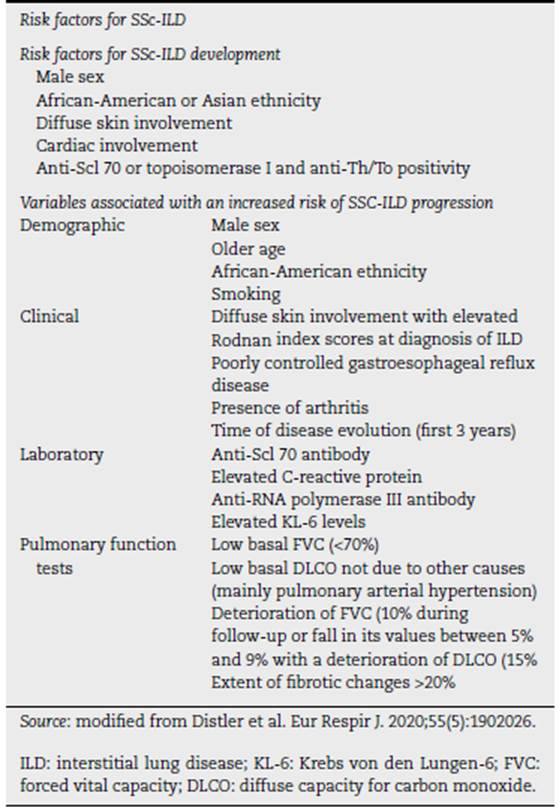
In Table 5, risk factors related to the development and progression of SSc-ILD are listed. (35
Table 5 Risk factors for systemic sclerosis related interstitial lung disease (SSc-ILD) development and progression.
Progression of ILD is characterized by deterioration in lung function tests, evaluated through forced vital capacity (FVC) and diffuse capacity for carbon monoxide (DLCO). In SSc-ILD, the European Trials and Research Group on Scleroderma (EUSTAR), since 2010, assessed the rate of progression in 12-month periods over 5 years. Of the cohort, 2259 (38%) patients had evidence of ILD by imaging, but only 826 were eligible for analysis. The course of the disease was evaluated as the change between the basal FVC and the last available. They found that 49 (9%) showed a significant decrease in FVC (decline >20%), 75 (14%) had a significant reduction (10-20%), 76 (14%) experienced a moderate decrease (5-10%), 206 (39%) remained stable (FVC changes <5%), and 129 (24%) experienced an improvement in FVC (>5%). Most patients with SSc-ILD had a pattern of slow decline in lung function, with a greater number of periods of stability or improvement than of worsening (58%); 34% showed a progressive course, with more worsening than stability or improvement periods; only 16 (8%) patients showed rapidly decreasing patterns of % of progression to PF.
FVC, with several consecutive episodes of worsening of FVC and absence of stable periods. (36 According to the recently proposed criteria for PPF, (37 27% would meet this diagnosis in the first year of follow-up (12% showed deterioration in FVC >10% and 15% decline in FVC >5%), which reveals considerable heterogeneity in the clinical course of SSc-ILD.
In the particular case of systemic sclerosis, the progression of the disease is defined as a decrease in FVC of ≥10%, or a reduction in FVC of 5-9% in combination with a decreased DLCO of ≥15%, as shown in Fig. 5. Therefore, it is essential to validate in this population the recently proposed criteria for progressive pulmonary fibrosis. (38
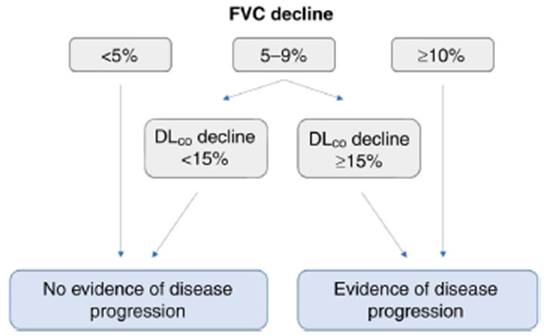
Source: modified from Ref. 38
Fig. 5 Proposed definition of disease progression based on FVC decline. FVC: forced vital capacity; DLCO: diffuse capacity for carbon monoxide.
The SPAR (SPO2 and ARthritis) model predicts progression in patients with mild SSc-ILD, defined as a relative decrease in FVC ≥15%, or FVC% ≥10%, combined with a decline in DLCO ≥15%, finding that oxygen saturation after a 6-minute walk less than 94% and the presence of arthritis at any time increase the likelihood of up to 86% of presenting worsening. (39
Biomarkers to predict the development of ILD and progression have been assessed with very interesting results. A study evaluated serum samples collected in the Scleroderma Lung Study II (a controlled and randomized clinical trial of mycophenolate mofetil (MMF) versus cyclophosphamide (CYC)) and validated in an independent observational cohort that was receiving treatment for SSc-ILD, calculated a composite score of six interferon-induced proteins (IFNγ inducible 10 kDa protein, IFN-y induced monocine, monocyte chemotactic protein 2, microglobulin β2, tumor necrosis factor receptor type II and macrophage inflammatory protein 3β) and its predictive value. A high initial score better predicted response to treatment with MMF (point estimate = 0.41, p = 0.001) and CYC (point assessment = 0.91, p = 0.009). Conversely, higher basal levels of C-reactive protein (PCR) predicted a worse course of ILD in both treatment arms. (40 A second study, conducted by Bowman et al., evaluated in an initial cohort of 385 patients and in a validation cohort of 204 patients, 31 biomarkers were associated with PPF in the derivation cohort, but only 17 were validated. The validated biomarkers showed a consistent association with PPF regardless of the cause of ILD. A proteomic signature comprising 12 biomarkers derived by artificial intelligence and validated in the University of Texas cohort showed a sensitivity of 0.90 and a negative predictive value of 0.91, suggesting that only 10% of patients with a low-risk protein signature would experience a progression of ILD in the following year. Those with a low-risk proteomic sign showed a change in FVC of +85.7 ml (95% CI 6.9-164.4), contrary to what was observed in high-risk sign subjects who had a change of FVC of -227.1ml (-286.7 to -167.5), this information is presumed to be helpful in planning future clinical trials and in decision-making for early initiation of treatment. (41
Treatment of progressive pulmonary fibrosis in SSc-ILD
Not all SSc-ILD patients require treatment; usually, those with extensive engagement or clinical ILD are initially handled with immunosuppression, while those who are not candidates to be treated should be closely monitored. The standard of care for clinical SSc-ILD is MMF, cyclophosphamide, azathioprine, or rituximab, supported by clinical trials that demonstrated stability in the decline of FVC values.
The INBUILD trial was a multicenter, double-blind, randomized clinical trial that evaluated the efficacy of nintedanib versus placebo in patients with non-IPF fibrosing lung disease with involvement of more than 10% of lung volume by HRCT. Patients met the criteria for lung disease progression despite treatment 24 months before randomization, a forced vital capacity (FVC) not less than 45% predicted, and DLCO between 30 and 80% of the predicted value. This trial included patients with CTD-ILD (which corresponded to 25% of the 663 patients studied), fulfilling the primary outcome, since the decrease in FVC was -80.8 ml/year with nintedanib and -187.8 ml/year with placebo for a statistically significant difference between groups of 107.0 ml/year (95% confidence interval [CI] 65.4-148.5; p< 0.001). Similar results were obtained when stratified by an ILD pattern similar to UIP, with an adjusted rate of FVC decrease of -82.9 ml/year with nintedanib and -211.1 ml/year with placebo, with a difference of 128.2ml (95% CI 70.8-185.6; p<0.001). Diarrhea was the most common adverse event, as reported in 66.9% and 23.9% of patients treated with nintedanib and placebo, respectively. (9 A post hoc analysis reported the response observed in the 170 patients with CTD-ILD, of which 89 had RA-ILD, 39 had SSc-ILD, 19 had MCTD-ILD, and 23 had ILD related to other autoimmune diseases. In the whole group, the rate of decrease in FVC at week 52 weeks was -75.9 ml/year with nintedanib versus -178.6 ml/year with placebo (difference 102.7 ml/year [95% CI 23.2,182.2]; nominal p = 0.012). This trial does not have sufficient power to identify statistically significant differences in each subgroup; in patients with SSc-ILD (39 out of 170) (the difference found was -120.7 ml/year [95% CI -53.2,294.6]; nominal p = 0.041) in favor of nintedanib treatment. (42
SENSCIS study randomly assigned SSc-ILD patients to receive either nintedanib 150 mg twice daily or a placebo. There were 576 participants (nintedanib n = 288 and placebo n = 288). Diarrhea was the most common adverse event, occurring in 75.7% of nintedanib patients and 31.6% of placebo patients; it led to permanent treatment discontinuation in 6.9% and 0.3% of nintedanib and placebo patients, respectively. Compared to placebo, treatment with nintedanib for 52 weeks resulted in a slower decline in FVC at 1 year. Approximately half of the patients received mycophenolate (48.3%); in a subgroup analysis, the effect of nintedanib treatment on the annual rate of decline in FVC was numerically greater in participants not taking mycophenolate at baseline (difference: 55.4ml/year [95% CI 2.3-108.5%]) than in those taking mycophenolate (26.3 ml/year [-27.9 to 80.6]). However, the relative effect of treatment with nintedanib was comparable between the two subgroups. Similar adverse events were observed in both subgroups, suggesting combining of mycophenolate and nintedanib is a safe treatment option for SSc-ILD patients. (43
We are currently awaiting the definitive results of the SLS III (Scleroderma Lung Study) study, which compares the efficacy and safety of pirfenidone, another approved antifibrotic for idiopathic pulmonary fibrosis, in combination with MMF versus MMF alone.
To conclude, one-third of patients with systemic sclerosis present a progression. PPF patients have a prognosis comparable to idiopathic pulmonary fibrosis patients. Once SSc is diagnosed, a systematic and active search for lung involvement must be conducted to achieve early detection and identify patients who will benefit from treatment. Patients at risk or with progression need immunosuppressive therapy; if progression starts or continues antifibrotic drugs (nintedanib) in combination with immunosuppression must be considered.
Progressive pulmonary fibrosis in RA-ILD
ILD is the most common pulmonary manifestation of RA. The prevalence differs between studies, but a prevalence as high as 60% has been reported. RA-ILD confers a poor prognosis, leading to high hospitalization rates, a low quality of life, and mortality, representing the second leading cause of mortality in RA after cardiovascular diseases. (24 A retrospective study with 1500 RA patients reported that the risk of death is twice as high in RA-ILD patients compared with AR patients without lung involvement. (23
UIP is the most common HRCT pattern in patients with RA-ILD. According to the PERSEIDS study, 60% of patients with RA-ILD have a UIP pattern10,44 which seems to confer the worst prognosis and a high risk of disease progression. (24 Other HRCT patterns may be seen in RA-ILD patients, especially NSIP. (23,44 The progression rate in RA-ILD is significant. Two retrospective European studies reported progression to fibrosis patterns between 38 and 50%.10,23 Fibrosis confers the worst prognosis and a high mortality rate. In a study conducted by Faverio et al. in two Italian centers, one-third of patients progressed despite treatment, with a mortality rate of 20% 3 years after diagnosis. (20
Risk factors, detection, and current assessment
Since a large number of people with RA develop pulmonary fibrosis, and the rate of progression is high in this group, it is vital to diagnose RA-ILD as soon as possible. Risk factors for RA-ILD have been described, such as smoking history, older age, male sex, long duration of disease, high articular activity, high levels of rheumatoid factor (RF) and citrulline peptide antibodies. (23,24 Chen et al. identified some risk factors for RA-ILD progression in 75 RA patients, with statistical significance in multivariate analysis, presented in Table 6.
Table 6 Risk factors for the progression of rheumatoid arthritis related interstitial lung disease (RA-ILD).

Another study that assessed clinical predictors of progressive pulmonary fibrosis in CTD-ILD (16% of patients had RA-ILD) found that a fibrotic pattern on HRCT at baseline, diabetes mellitus, and steroid use have a higher risk of developing PPF. This condition is linked to higher mortality in this subgroup of patients. (45
Diagnostic methods for RA-ILD are not systematically standardized; however, many physicians use similar approaches to SS-ILD. In the study by Takizawa et al., 574 pulmonologists, rheumatologists, and internal medicine physicians filled out an online survey about how ILD, including PPF, is currently diagnosed and treated. Concerning CTD-ILD, the most common tests used were high-resolution tomography (HRCT), pulmonary function tests (spirometry, DLCO), and the 6-minute walk test. Other tests, such as biomarkers and pulso-oximetry, were also on the list. The frequency of which tests are performed varies between regions. (6
Biomarkers
There are already biomarkers related to CTD-ILD and progression to fibrosis, such as CXCL13, CA-125, MMP7, YKL-40, SP-D, VCAM and KL-6. (41 Bowman et al. identified and validated 17 plasma biomarkers associated with PPF independently of the etiology. A proteomic signature with 12 biomarkers was developed; patients with a "high risk" proteomic signature had more decline in FVC in the next year versus "low-risk patients" (85.7 ml vs. 227.1 ml, respectively), and patients with the high-risk proteomic signature had an almost seven-fold higher risk of progressive fibrosing ILD compared with the low-risk signature (OR 6.73, 95% CI 4.00-11.33). This study was done with a validation cohort of 204 ILD patients (21 with RA) and a discovery cohort of 385 ILD patients (38 with RA). (41
Most of these biomarkers have epithelial and mesenchymal cell origins. ITGB6 was the biomarker with the strongest association with progressive fibrosing ILD. Table 7 shows the biomarker signature proposed by the authors and their associations with the progression to fibrosis in patients with CTD-ILD in the cohort.
Table 7 Biomarker signature and the association to progressive fibrosis in the 235 connective tissue disease-related interstitial lung disease (CTD-ILD) patients of the cohort.
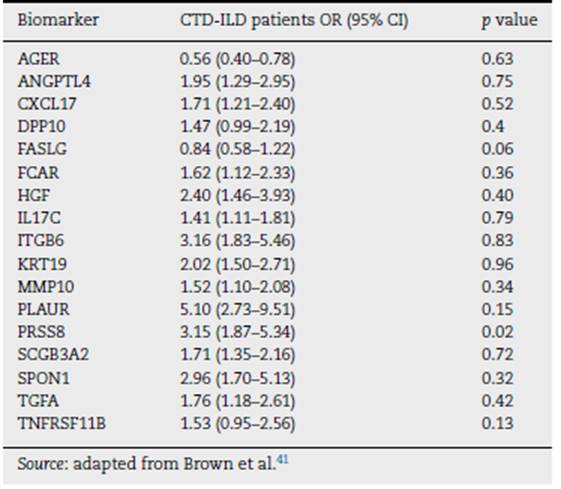
Even though this is a promising tool to identifying ILD patients at risk of progressing to PPF, further studies are needed to identify their impact in the clinical setting. This study included CTD-ILD patients, some with RA, but there was no a specific biomarker related to RA-ILD.
Treatment of progressive pulmonary fibrosis in RA-ILD
Control of activity-related disease has been shown to have a positive effect on controlling RA-ILD. (23 Some immunosuppressive drugs, like abatacept, rituximab, tocilizumab, and Janus kinase (JAK) inhibitors, work just as well in the articular domain as they do in the pulmonary domain. (46-49 Despite this treatment some patients will progress, and other treatment options need to be considered.
Of 663 patients in the INBUILD trial, 89 (13.4%) had RA-ILD. The baseline characteristics of the patients were mean (SD) age was 66.9 (9.6) years, 60.7% were male, and 86.5% had a UIP-like fibrotic pattern on HRCT. Mean (SD) FVC was 71.5 (16.2)% predicted, and mean (SD) DLCO was 47.7 (15.6)% predicted. Mean (SD) times since diagnosis of RA and RA-ILD were 9.9 (9.4) years and 3.6 (3.2) years. Among patients with available CRP measurements, the mean (SD) hs-CRP was 13.7 (22.5) mg/L. 21.3% took biologic DMARDs, 53.9% non-biologic DMARDs, and 73.0% glucocorticoids. The rate of decline in FVC at week 52 was -82.6 ml in the nintedanib group versus -199.3 ml/year in the nintedanib group. Placebo (difference 116.7 ml/year (95% CI 7.4,226.1), nominal p = 0.037. The most common adverse event was diarrhea, reported by 61.9% and 27.7% of the nintedanib and placebo groups led to drug discontinuation in 23.8% and 17% of subjects in the nintedanib and placebo groups. These data from the INBUILD trial show that nintedanib reduced the rate of decline in FVC over 52 weeks in patients with progres sive fibrosing RA-ILD by 59% compared with placebo, similar to the relative treatment effect observed in the overall trial population, (15 in patients with SSc-ILD20 and in patients with IPF. (54
In a real-life multicenter retrospective study, the medical records of people with PPF were reviewed. Sixty-four patients had IPF, and 103 had non-IPF; in the last group, 31 patients had CTD-ILD, and 8 patients had RA, representing 7.7% of the non-IPF patients. In the non-IPF patients, 34 were on antifibrotic treatment and 69 were not. Overall survival and the rate of FCV decline were measured in both groups. The study reported that antifibrotics reduced the rate of FCV decline in IPF and non-IPF patients, but differences in mortality between patients with and without antifibrotic treatment were not statistically significant in the last group of patients. (50 Further studies are needed to evaluate the impact of reducing the rate of FVC decline on outcomes such as mortality.
Finally, TRAIL1 was a randomized, double-blind, placebo-controlled, phase 2 trial conducted in four countries (the UK, the USA, Australia, and Canada). Adults aged 18 85 years were eligible for inclusion if they met the 2010 American College of Rheumatology and European Alliance of Associations for Rheumatology criteria for rheumatoid arthritis and had interstitial lung disease on a high-resolution CT scan imaging and, when available, lung biopsy. Patients were randomly given 2403 mg oral pirfenidone (pirfenidone group) or placebo (placebo group) daily. The primary endpoint was the incidence of the composite endpoint of a decline from baseline in percent predicted forced vital capacity (FVC%) of 10% or more or death during the 52-week treatment period. Key secondary endpoints included change in absolute and FVC% over 52 weeks and the proportion of patients with a decline in FVC% of 10% or more. The difference in the proportion of patients who met the composite primary endpoint between the two groups was not significant (seven [11%] of 63 patients in the pirfenidone group vs. nine [15%] of 60 patients in the placebo group); OR 0.67 (95% CI 0.22 to 2.03); p = 0.48. Compared with the placebo group, patients in the pirfenidone group had a slower rate of decline in lung function, measured by the estimated annual change in absolute FVC (-66 vs. -146; p = 0.0082). The groups were similar with regards to the decline in FVC% by 10% or more five (8%) participants in the pirfenidone group versus seven (12%) in the placebo group; OR 0.52 (95% CI 0.14-1.90); p = 0.32. There was no significant difference in the treatment-emergent serious adverse events rate between the two groups and no treatment-related deaths. Due to the early termination of the study and underpowering, the results should be interpreted with caution. Despite not meeting the composite primary endpoint, pirfenidone slowed the rate of decline of FVC over time in patients with RA-ILD. Safety in patients with RA-ILD was similar to that seen in other pirfenidone trials. (55
PPF associated with other CTD-ILDs (Sjogren, MCTD, UCTD, ANCA-associated, SLE, IIM)
Interstitial lung involvement in autoimmune pathology is common, as stated in the preceding paragraphs. (1,5 Nagy et al. documented a ILD prevalence of 50.8% for SSc, 20.6% for RA, 9.5% for SLE, 9.5% for others (MCTD and UCTD), 6.4% for IIM, and 3.2% for vasculitis. (26 CTD has been identified as a subgroup of pathologies that can lead to a progressive fibrosant pattern unrelated to idiopathic pulmonary fibrosis; consequently, it has been the subject of numerous epidemiological, historical, prospective, and real-world studies. (45 AR and SSc have been identified as the primary causes of the progressive pattern, (26,50-52 whereas Sjogren syndrome, MCTD, UCTD, IIM, and SLE account for a smaller proportion of cases. (12,13 Even in the largest clinical trials or observational studies, insufficient data are available for these pathologies, (53 limiting the ability to analyze the epidemiology, risks, and treatment of PPF in diseases other than RA and SSc.
Conclusion
Progressive pulmonary fibrosis is a condition that confers worst prognosis in patients with CTD-ILD. Data suggest that has a relevant prevalence in SSc and RA and is related with high mortality rates. It is important to diagnose this condition as soon as possible to stablish treatment that may impact in the disease course.
Immunosuppressive therapy has shown to control the disease-related activity, but some patients may progress despite this treatment. In this cases antifibrotics need to be considered.
Other CTD as Sjogren syndrome, inflammatory myopathies, MTCT and UCTD are related with ILD and progressive fibrosis. There is not enough evidence to stablish the prevalence and characteristics of this condition in this population, data is usually extra polled from the RA-ILD and SSC-ILD studies.

