










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
Print version ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.36 no.140 Bogotá July/Sept. 2012
QUÍMICA
*Â Grupo de Química Ambiental y Computacional. Universidad de Cartagena, Colombia. Email: joliverov@unicartagena.edu.co; jesusolivero@yahoo.com.
RESUMEN
El resurgimiento de los productos naturales en el ámbito del descubrimiento de fármacos tiene su base en la introducción de nuevos enfoques, tecnologías y metodologías, incluyendo el diseño de moléculas bioactivas asistido por ordenador. Los servidores TarFisDock y PharmMapper son herramientas computacionales empleadas para la búsqueda de potenciales blancos farmacológicos para ligandos orgánicos. Este trabajo muestra un ejemplo de aplicación de estos servidores. Los resultados obtenidos sugirieron que moléculas de origen natural, tales como la ascididemina (Didemnum sp. Tunicado), el peyssonol B (Peyssonnelia sp), el ácido asiático (Centella asiatica) y el ácido docosahexaenoico (aceite de pescado), tienen como blancos la dihidroorotato deshidrogenasa, la mieloperoxidasa, el receptor beta del ácido retinoico, y la aldosa reductasa, respectivamente. Estas proteínas han sido relacionadas con diversas enfermedades y ofrecen una explicación teórica de algunos de los efectos benéficos reportados para las moléculas evaluadas.
Palabras clave: AutoDock Vina, productos naturales, PharmMapper, farmacóforo, TarFisDock, acoplamiento molecular.
ABSTRACT
The resurgence of natural products on the drug discovery scene has its basis on the introduction of new approaches, technologies and methodologies, including the computer-aided design of bioactive molecules. TarFisDock and PharmMapper severs are computational tools employed for the search of potential drug targets for organic ligands. This work shows an example of application for these servers. The obtained results suggested that nature-derived molecules, such as ascididemine (Didemnum sp. Tunicado), peyssonol B (Peyssonnelia sp), asiatic acid (Centella asiatica) and the docosahexaenoic acid (fish oil), have as targets the dihydroorotate dehydrogenase, myeloperoxidase, retinoic acid receptor beta, and aldose reductase, respectivaley. These proteins have been related to diverse diseases and offer a theoretical explanation for some of the beneficial effects reported for evaluated molecules.
Keywords: AutoDock Vina, natural products, PharmMapper, pharmacophore, TarFisDock, Molecular docking.
Introducción
Muchas revisiones científicas han documentado la utilización de los recursos naturales, plantas, animales y microorganismos, entre otros, con fines medicinales (Ji et al., 2009; Ganesan, 2008; Butler, 2006; Rishton, 2006), práctica que ha estado presente desde el principio de la humanidad (Ji et al., 2009), constituyéndose en la principal fuente de ingredientes activos empleados en la industria farmacéutica (Harvey et al., 2008).
Las interacciones que tienen lugar entre los organismos en los ecosistemas les ha ayudado a adaptarse a su hábitat. Este proceso ha dado lugar a una amplia variedad de compuestos naturales complejos, los cuales pueden interactuar de manera específica con moléculas biológicas blanco (McChesney et al., 2007). Con las millones de estructuras diferentes resultantes de la química combinatoria natural, ha sido estimado que la diversidad de los compuestos naturales es mayor que la colección de productos de síntesis que existen en la actualidad (Wilk et al., 2010; Kumar & Waldmann, 2009; Harvey, 2008). Otra de las ventajas de los compuestos naturales sobre los fármacos sintéticos radica en la mayor similitud de sus estructuras con el "espacio químico" ocupado en las proteínas objetivos, permitiendo una mejor acción sobre estas (Harvey, 2007).
El papel de las moléculas de origen natural es notable en la producción de fármacos beneficiosos para la salud humana. Más del 50% de los medicamentos aprobados desde 1994 derivan de ingredientes naturales (Mc Chesney et al., 2007). Entre los múltiples ejemplos están los medicamentos usados para combatir el cáncer y diferentes tipos de infección (McChesney et al., 2007; Lam, 2007; Cordell, 2000). Desafortunadamente, desde la década de 1990's, muchas compañías farmacéuticas redujeron la búsqueda y el interés en el desarrollo de nuevos medicamentos a partir de compuestos naturales (Harvey, 2008; Lam, 2007). Esto sumado a la aparición de tecnologías como la búsqueda de alto rendimiento (High Throughput Screening, HTS), acoplada con la química combinatoria, originó recortes en la investigación en esta área. La meta era acelerar el tiempo de desarrollo de nuevos medicamentos y evitar las dificultades derivadas de la utilización de productos naturales, incluyendo el acceso y el suministro de los mismos y su complejidad química (Harvey, 2008; Lam, 2007). Sin embargo, la disponibilidad estructural de biomoléculas, la aparición de herramientas de acoplamiento proteína-ligando y el desarrollo de metodologías de diseño asistido por computadores (Mandal et al., 2010), han facilitado el resurgimiento del interés en el descubrimiento de nuevos fármacos a partir de productos naturales.
La búsqueda, el diseño y la optimización de nuevas moléculas con actividad biológica específica, ha conducido a que la industria farmacéutica sea una de las líderes en este campo de la investigación in silico (Wild & Wiggins, 2006). Dentro de las herramientas computacionales más comunes en esta área están el diseño basado en la estructura del blanco molecular (acoplamiento inverso proteína-ligando), en el ligando (farmacóforo), y en las relaciones cuantitativas estructura-actividad/ propiedad (QSAR/QSPR), para la predicción de la actividad biológica de forma computacional (Kapetanovic, 2008).
Acoplamiento inverso proteína-ligando
Los métodos tradicionales de acoplamiento proteína-ligando en general unen moléculas flexibles dentro de una representación específica y rígida del receptor (proteína), lo cual puede introducir errores en los cálculos convirtiéndose en desventajas (Harriman et al., 2007). Hoy en día, los programas de mayor uso realizan el acoplamiento permitiendo flexibilidad en los residuos del sitio de unión a la proteína y en los ligandos, proporcionando de esta manera simulaciones más realistas.
En el acoplamiento inverso o reverso, el receptor es tratado como un huésped, y el proceso es realizado alrededor de un ligando rígido (Kämper et al., 2006; Harriman et al., 2007). Este enfoque es utilizado principalmente en la búsqueda de posibles proteínas blanco para moléculas pequeñas con actividad biológica conocida, empleando métodos de acoplamiento entre las estructuras almacenadas en una base de datos de proteínas con cavidad y un ligando específico (Li et al., 2006). Los complejos con la mejor interacción proteína-ligando permiten identificar candidatos potenciales para la validación experimental (Tang et al., 2006).
Entre las ventajas y posibles aplicaciones del método de acoplamiento inverso dirigidas al descubrimiento de fármacos, existen varias contribuciones importantes, incluyendo la detección de blancos terapéuticos desconocidos, la identificación de blancos potenciales responsables de la toxicidad y/o efectos secundarios de un medicamento, así como la exploración de mecanismos moleculares de compuestos bioactivos a base de plantas, en particular aquellas provenientes de las medicinas tradicionales (Mandal et al., 2010; Tang et al., 2006; Chen & Ung, 2002).
En la actualidad hay varios servidores y softwares utilizados para el acoplamiento inverso, tales como INVDOCK (Chen & Ung, 2002), EM-Dock 3 (Harriman et al., 2007), y TarFisDock (Li et al., 2006), entre otros.
TarFisDock
El servidor TarFisDock (Target Fishing Dock) es una herramienta disponible en la web creada por el Centro de Diseño y Descubrimiento de Fármacos (DDDC) de Shanghai, China, y desarrollada sobre la base del programa de acoplamiento Dock 4.0 (Li et al., 2006). Tiene como propósito encontrar proteínas que posiblemente sirvan de blanco para pequeñas moléculas activas que fungen como ligandos (Cai et al.,2006). Es posible acceder libremente a esta herramienta a través de la página web http://www.dddc.ac.cn/tarfisdock/.
TarFisDock está integrado con una base de datos de blancos farmacológicos potenciales (PDTD), la cual funciona como piedra angular para identificar los posibles blancos enlazantes in silico (Gao et al., 2008). Esta colección de proteínas fue construida a partir de la literatura, y bases de datos en línea tales como DrugBank, Base de Datos de Blancos Terapéuticos (TTD), y Thomson Pharma (Tang et al., 2006; Li et al., 2006, Gao et al., 2008).
De acuerdo con el sitio web de PDTD (http://www.dddc.ac.cn/pdtd/), en la actualidad (2012), esta base de datos contiene 1207 entradas con estructuras 3D de la base de datos Protein Data Bank (PDB, http://www.pdb.org/pdb/home/home.do), las cuales pueden ser evaluadas como blancos. Las proteínas o blancos farmacológicos están clasificadas de acuerdo con dos criterios: áreas terapéuticas y función bioquímica.
La base de datos PDTD tiene diferentes funciones de búsqueda. Los usuarios pueden acceder a datos por el código PDB, nombre de la proteína o nombre de la enfermedad. Resultados de la consulta se presentan en un formato de tabla y cada proteína blanco tiene su propia página de resultados para proporcionar información comprensible, incluyendo el código PDB, nombre, estructura, categoría, enfermedad relacionada, y el sitio activo de la proteína (Gao et al., 2008). Además, ofrece enlaces externos a otras bases de datos asociadas, como Therapeutic Target Database (TTD), DrugBank, el servidor de proteómica ExPASy, La Enciclopedia de Genes y Genomas de Kyoto (KEGG), entre otras (Mattingly, 2009; Gao et al., 2008). La estructura de la pequeña molécula analizada debe ser presentada en formato Mol2 (Li et al., 2006).
A pesar de todo su potencial, TarFisDock tiene algunas limitaciones. Por ejemplo, no contempla la flexibilidad de las proteínas durante la simulación de acoplamiento, y las disponibles en su base de datos solo cubren una pequeña fracción de proteínas que han sido relacionadas con enfermedades comunes (Li et al., 2006). Este hecho es reconocido por sus propios creadores y se encuentra en un proceso de mejora continua.
Mapeo del farmacóforo
Una parte esencial de la búsqueda y diseño de fármacos es la predicción del posible acoplamiento o interacción entre moléculas pequeñas y macromoléculas blanco. Este proceso puede ser realizado empleando el concepto de farmacóforo. De acuerdo con la definición de la IUPAC, un farmacóforo es "un conjunto de características estéricas y electrónicas que es necesario para asegurar la óptima interacción supramolecular con un blanco biológico específico y para activar (o bloquear) su respuesta biológica" (Wermuth et al., 1998). El concepto farmacóforo se basa en los tipos de interacción observadas en el reconocimiento molecular, tales como enlaces de hidrógeno, la carga positiva o negativa y regiones hidrofóbicas (por ejemplo, centros de anillos aromáticos) (Mukherjee et al., 2008). El mapeo del farmacóforo es una de las herramientas computacionales más exitosas y un paso para entender el proceso de reconocimiento ligando-receptor (Kansal et al., 2010).
Los modelos de farmacóforos son utilizados cuando un compuesto activo ha sido identificado, siendo la estructura 3D de la proteína blanco o receptor por lo general desconocida (Gab, 2008). Los farmacóforos intentan proveer una matriz de tres dimensiones de los grupos funcionales que una molécula debe poseer para ser reconocido por el receptor (Kansal et al., 2010). Un mapeo de farmacóforo es una descripción 3D del mismo, desarrollado mediante la especificación de la naturaleza de las características electrónicas y topológicas claves, así como de su distancia 3D en el mapa (Gad, 2008).
Un uso corriente del farmacóforo es explorar bases de datos que los contienen, identificando de esta forma nuevas clases estructurales de compuestos potenciales activos (Jain & Chincholikar, 2004). Para tal efecto, existen varios programas disponibles en el comercio, por ejemplo CATALYST, GASP y DISCO (Mukherjee et al., 2008), así como servidores en la web tales como PharmaGist (Schneidman-Du hovny et al., 2008). Estas plataformas son utilizadas para la generación de farmacóforos para una o varias moléculas. Sin embargo, recientemente existen nuevas herramientas poderosas en la web que permiten realizar enfoques basados en modelos de farmacóforos para encontrar proteínas blanco por acoplamiento inverso, un ejemplo destacado es el nuevo servidor llamado PharmMapper.
PharmMapper
PharmMapper es una herramienta de servidor web puesta en funcionamiento en 2009 por el Centro de Diseño y Descubrimiento de Fármacos (DDDC) de Shanghai. El propósito de esta herramienta es identificar blancos potenciales de pequeñas moléculas (medicamentos, productos naturales u otros compuestos con blancos enlazantes no identificados) con un mapeo "inverso" del farmacóforo (Liu et al., 2010). PharmMapper cuenta con una colección de farmacóforos, en una base de datos llamada "PharmTargetDB". Estos farmacóforos fueron extraídos de blancos presentes en TargetBank, DrugBank, BindingDB y PDTD. Ademas, contiene anotaciones obtenidas de DrugBank, PDBSum, UniProt y TargetBank. En el proceso de derivación del farmacóforo, PharmMapper emplea el software Ligandscout (Wolber & Langer, 2005)
Según su página web (http://59.78.96.61/pharmmapper/), PharmMapper permite acceder a más de 7000 modelos de farmacóforos que cubren 1627 proteinas blanco de interes farmacéutico, 459 de las cuales son humanas. PharmMapper hace una búsqueda en la base de datos PharmTargetDB y encuentra la mejor asignación de poses conformacionales de la molécula evaluada en todos los modelos de farmacóforo en su base de datos, generando un listado de los mejores blancos con sus anotaciones correspondientes (Liu et al., 2010). El número de estructuras de PharmTargetDB es limitado, pero aumenta de forma constante, siendo claro que PharmMaper posee un gran potencial en el campo del descubrimiento de fármacos.
Varios estudios han sido llevados a cabo utilizando TarFisDock y PharmMapper, como herramientas en la identificación de blancos terapéuticos. Cai et al (2006), han demostrado el valor potencial de TarFisDock para la identificación de la proteína blanco de un producto natural, N-trans-caffeoiltiramina, en el proteoma del Helicobacter pyroli. Dos de las quince proteínas sugeridas como blancos por TarFisDock, tenían secuencias homólogas en H. pylori: la diaminopimelato descarboxilasa (DC) y la péptido deformilasa (PDF). Después de la validación enzimática, sólo la proteína PDF resultó ser un blanco probable. La estructura cristalina del complejo N-trans-caffeoiltiramina/PDF sugiere un enlace muy selectivo en el sitio de unión con esta proteína.
Pandey et al en 2009 síntetizaron una biblioteca de análogos de "carba" péptidos, tomando como plataforma de diseño el ácido glicosilamino. Mediante la utilización de PDTD, los autores identificaron dos blancos potenciales de estos compuestos: la proteína codificada por el gen lysA y la dihidropicolinato reductasa de M. tuberculosis. Ambos hacen parte de la ruta biosintética de la lisina y están ausentes en los seres humanos, por lo que podrían ser sugeridos para el control de la tuberculosis. Jeong et al (2009), mediante el uso de TarFisDock, encontraron que la Leucotrieno A4 hidrolasa (LTA4H) podría ser un blanco potencial de [6]-gingerol, un compuesto natural de jengibre, que muestra actividad antiinflamatoria y anti-tumorigénica. Estos autores demostraron experimentalmente que el [6]-gingerol suprime el crecimiento tumoral in vivo, efecto mediado por la inhibición de la actividad de LTA4H.
En el año 2010, Olivero et al., utilizaron TarFisDock para hacer un cribado virtual con el fin de encontrar las proteínas diana de la TCDD (2,3,7,8-tetraclorodibenzo-p-dioxina), un compuesto altamente tóxico. Los autores concluyeron que, al menos teóricamente, existen varias proteínas con alta afinidad para la TCDD, tales como las metalopeptidasas 8 y 3, la oxidoescualeno ciclasa, y la mieloperoxidasa, lo cual implicaría que el TCDD puede actuar por mecanismos diferentes al mediado por el receptor de hidrocarburos, el cual es su blanco conocido.
Para el año siguiente (2011) Maldonado et al., usando el servidor TarFisDock, identificaron posibles blancos farmacológicos para el ácido alfa lipoico (R-ALA). Los resultados in silico sugierieron que el R-ALA puede interaccionar con enzimas presentes en mamíferos, tales como la LTA4H, el canal de potasio dependiente de voltaje, la alfa hidroxiesteroide deshidrogenasa, y la epóxido hidrolasa, proteínas implicadas en enfermedades como el cáncer, diabetes, trastornos neurológicos y cardiovasculares. Chen et al., (2011) emplearon el servidor PharmMapper para la identificación de blancos farmacológicos potenciales para el compuesto etil-8-oxo-8H-acenafto[1,2-b]pirrol-9-carboxilato, usando la base de datos PharmTargetDB, identificaron dos enzimas tirosina quinasas (LCK y Src) y tres serina/treonina quinasas (MEK1, CDK2 y p38 MAPK) que interaccionaban con este compuesto, hecho que fue igualmente corroborado experimentalmente.
Ejercicio de aplicación con TarFisDock y PharmMapper
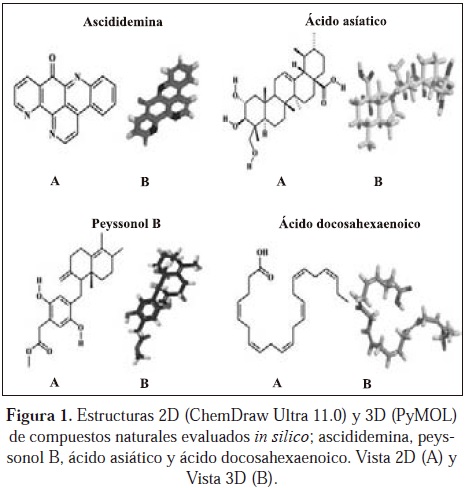
Con el propósito de explorar las capacidades de los servidores TarfisDock y PharmMapper, y evidenciar su potencial como herramientas computacionales para encontrar nuevas blancos farmacológicos de compuestos naturales, cuatro moléculas con actividad biológica reportada fueron utilizadas: el ácido asiático de la planta Centella asiática (Patil, et al., 2010), el ácido docosahexaenoico, un ácido graso omega-3 del aceite de pescado (Mullen et al., 2010), y el producto marino natural ascididemina, un alcaloide tipo piridoacridina, originalmente derivado del organismo tunicado Didemnum sp. (Matsumoto et al., 2003; Appleton et al., 2010).
Materiales y métodos
Modelado molecular de los compuestos naturales bioactivos
Un enfoque de tres pasos ha sido aplicado para identificar las proteínas que son blanco para el ácido asiático, el peyssonol B, el ácido docosahexaenoico, y la ascididemina (Fig. 1). El proceso incluye la optimización de los ligandos, el cribado virtual de nuevos blancos y, finalmente, el cálculo de la afinidad proteína-ligando. En primer lugar, las estructuras de los ligandos fueron optimizadas con el método de la teoría funcional de la densidad (DFT) a un nivel B3LYP/6-31G. Los cálculos fueron realizados con el programa Gaussian 03 (Frisch et al., 2003). La geometría resultante fue convertida a formato Mol2 con el programa Open Babel (Guha et al., 2006).
Identificación de proteínas diana de compuestos naturales bioactivos utilizando los servidores TarFisDock y PharmMapper.
Las cuatro estructuras optimizadas fueron enviadas al servidor TarFisDock para encontrar proteínas con estructuras tridimensionales con sitios teóricos de unión para cada uno de los compuestos estudiados. La búsqueda comenzó usando la opción "Targets in all categories", y el acoplamiento con cada ligando fue realizado en todas las proteínas presentes en PDTD. El resultado consiste en los mejores blancos potenciales clasificados por una puntación de energía (energy score), proporcionando conformaciones de interacción proteína-ligando para cada molécula evaluada, así como una tabla con información relacionada con los blancos farmacológicos. Los sitios de unión para aquellas proteínas presentes en mamíferos, correspondientes a los mejores 25 blancos farmacológicos sugeridos por TarFisDock, fueron analizados y verificados mediante el algoritmo de identificación de sitios (SiteID), incorporado en Sybyl 8.1.1 (Tripos, St. Louis, 2007).
Estos compuestos naturales bioactivos también fueron ingresadas al servidor PharmMapper, para encontrar los sitios de unión a proteínas que fuesen blancos teóricos potenciales. La búsqueda fue ejecutada empleando la opción "Targets in all categories", y la identificación de los posibles candidatos fue llevada a cabo con la técnica de mapeo del farmacóforo (7302 modelos de farmacóforo están disponibles en PharmTargetDB y 2241 de ellos aparecen anotados como proteínas provenientes de humanos). El resultado final consiste en los mejores blancos farmacológicos clasificados según una puntuación de ajuste (Fit Score), proporcionando las mejores poses (en formato Mol2) de la molécula evaluada sobre los candidatos farmacóforicos. Aquellas proteínas presentes en mamíferos y que fueron obtenidas de los mejores 25 blancos clasificados por PharmMapper, se analizaron y verificaron de la misma forma realizada con TarFisDock.
Acoplamiento de compuestos bioactivos naturales en las proteínas blanco.
Los archivos de salida de TarFisDock y PharmMapper para los compuestos bioactivos naturales examinados fueron acoplados a cada una de las proteínas seleccionadas (archivo 3D tomados de Protein Data Bank, www.pdb.org/pdb/home/home.do home/home.do) para su análisis. El cálculo de los valores de afinidad (kcal/mol) fue realizado utilizando el programa AutoDock Vina 1.0 (Trott & Olson, 2010). El sitio de acoplamiento para las estructuras de los ligandos fue definido estableciendo un cubo con las dimensiones 24× 24 × 24 Å, cubriendo el sitio de unión predicho por TarFisDock y PharMapper, con un espaciado de malla de 0.375 Å, fijado en el centro geométrico de cada ligando evaluado.
Identificación de los principales residuos que interactúan con los compuestos naturales bioactivos en el sitio de unión con proteínas blanco.
La identificación de residuos de proteínas que interaccionan en el sitio de unión con los compuestos naturales bioactivos para las proteínas que tuvieron el mejor valor de afinidad AutoDock Vina (kcal/mol) fue realizada con el programa LigandScout 3.0 (Wolber & Langer, 2005). Esta herramienta crea modelos de farmacóforos simplificados para detectar el número y tipo de interacción entre el ligando y los residuos existentes en el sitio de unión con la proteína. La visualización de las imágenes de cada complejo proteína-ligando fue realizada usando el programa PyMOL (DeLano, 2003).
Resultados y discusión
Los servidores TarFisDock y PharmMapper fueron utilizados para encontrar las proteínas diana de el ácido asiático, el peyssonol B, el ácido docosahexaenoico, y la ascididemina. AutoDock Vina 1.0 fue usado para el cálculo del valor teórico para la afinidad del acoplamiento de estos compuestos con las proteínas presentes en mamíferos, y que fueron obtenidas a partir de aquellas con los 25 mejores valores de puntuación generados por los servidores. Los tres mejores blancos farmacológicos predichos por TarFisDock y PharmMapper son presentados en las Tablas 1 y 2, respectivamente.
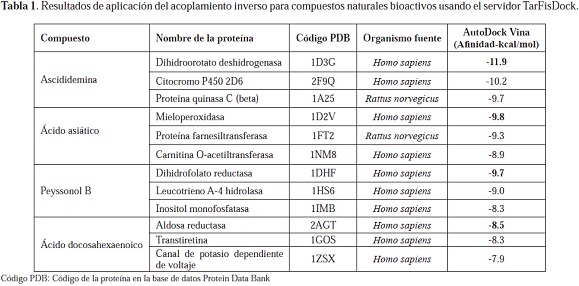
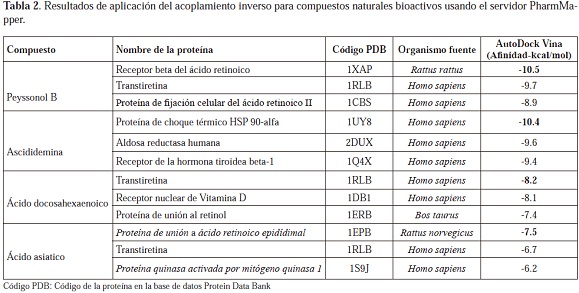
El cribado virtual de proteínas blanco para compuestos naturales bioactivos con TarFisDock y PharmMapper demostró que, al menos teóricamente, los cuatro compuestos evaluados podrían interaccionar con diferentes enzimas tales como: reductasas, isomerasas, hidrolasas, transferasas, quinasas y receptores moleculares, entre otras. Algunas de las proteínas predichas como blancos se encuentran en los mamíferos, y estos representan importantes dianas farmacológicas, principalmente aquellas que presentaron los valores absolutos más altos, en particular la dihidroorotato deshidrogenasa (PDB: 1D3G), el receptor beta del ácido retinoico (PDB: 1XAP), la mieloperoxidasa (PDB: 1D2V) y la aldosa reductasa (PDB: 2AGT), las cuales podrían ser blancos importantes para la ascididemina, el peyssonol B, el ácido asiático y el ácido docosahexaenoico, respectivamente.
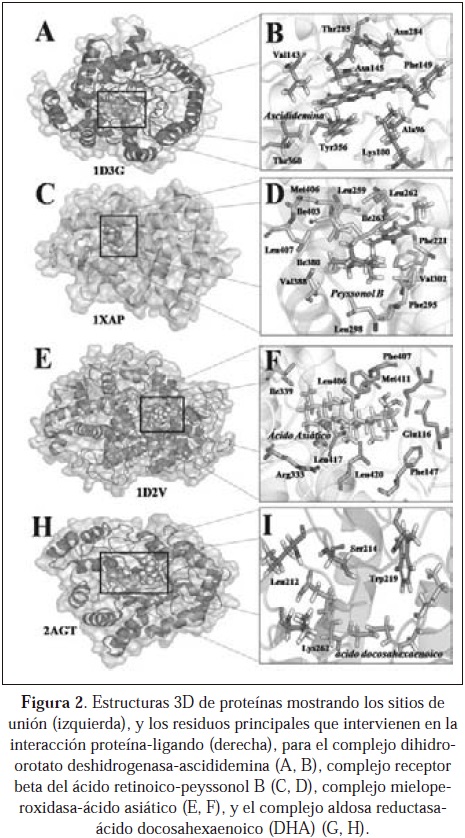
Los sitios de unión del ligando con estas proteínas, así como las interacciones entre sus residuos y cada uno de los compuestos naturales en cada complejo proteína-ligando, identificadas por LigandScout 3.0 son presentados en la Fig. 2. Las interacciones para el complejo dihidroorotato deshidrogenasa-ascididemina están determinadas por los residuos Tyr356 (anillo aromático e hidrofóbicas), Asn284, Lys100 (hidrógeno aceptor), Asn145 (hidrógeno dador) Val143, Phe149, Ala96, Thr285, y Thr360 (hidrofóbicas). Para el complejo receptor beta del ácido retinoico-peyssonol B, estos aminoácidos fueron Leu262 (hidrógeno dador), Phe221, Leu259, Ile263, Phe295, Leu298, Val302, Ile380, Val388, Ile403, Met406 y Leu407 (hidrofóbicas). Para el complejo mieloperoxidasa-ácido asiático los aminoácidos más importantes en el sitio de unión fueron Glu116 (hidrógeno dador), Phe147, Arg333, Ile339, Leu406, Phe407, Met411, Leu417, y Leu420 (hidrofóbicas). Por último, el complejo aldosa reductasa-ácido docosahexaenoico muestra un menor número de aminoácidos que interacionan en el sitio de unión con el ligando, estos fueron Leu212, Ser214, Lys262 (hidrógeno dador), y Trp219 (hidrofóbica).
Las proteínas predichas por los servidores TarFisDock y PharmMapper para los compuestos naturales bioactivos examinados en este ejemplo, constituyen mediadores bioquímicos de especial interés en varias vías conocidas de señalización molecular. La enzima dihidroorotato deshidrogenasa cataliza la biosíntesis de pirimidina mediante la conversión de dihidroorotato a orotato. Además, está implicada en enfermedades autoinmunes como el lupus eritematoso (Kulkarni et al., 2010), lo que la ha convertido en un importante blanco de los fármacos antiparasitarios y citostáticos (Zameitat et al., 2007).
El receptor beta del ácido retinoico, el cual interacciona con el ácido retinoico, la forma biológicamente activa de vitamina A, ha sido relacionado con enfermedades cardiovasculares, ya que su inhibición impide la formación de neoíntima, una capa engrosada de la íntima arterial, presente especialmente en aterosclerosis o implantación de prótesis, producida por migración y proliferación de células de la capa media de la intima (Kada et al., 2007). Por otra parte, este receptor ha sido considerado como un nuevo blanco terapéutico en la enfermedad de Alzheimer (Wolfe & Selkoe, 2010).
La mieloperoxidasa es una enzima expresada en neutrófilos humanos. Los estudios clínicos muestran que esta proteína puede aumentar la vulnerabilidad a la fibrilación auricular (Rudolph et al., 2010). Algunos estudios han reportado que los niveles séricos de mieloperoxidasa podrían predecir la disfunción endotelial y eventos vasculopáticos en humanos (Kaneski et al., 2006; Vita et al., 2004). Por otra parte, la acción catalítica de la aldosa reductasa ha sido asociada con la aparición de cataratas intumescentes, glaucoma secundario e inflamación ocular en pacientes diabéticos (Chomanska et al., 2010; Yadav et al., 2010). Además, estudios han relacionado esta enzima con algunos tipos de cáncer de colon en humanos (Ramana et al., 2010).
Aunque todavía hay mucho por descubrir sobre el mecanismo de acción de los compuestos naturales con actividad biológica, es muy importante indagar acerca de su acción a nivel de proteínas con el fin de tener un conocimiento más preciso que explique las principales causas de sus efectos benéficos y secundarios. La mayoría de los compuestos naturales han servido como terapia de base y plataforma para el diseño de nuevos fármacos. El Acoplamiento molecular inverso está siendo utilizado como una herramienta poderosa en estudios de química computacional encaminados a la búsqueda de nuevos fármacos, gracias a la velocidad y el bajo costo de los procesos, facilitando el arduo camino para llegar a la fase de experimentación, con argumentos más claros de acuerdo con los resultados teóricos obtenidos.
Los datos presentados en este trabajo, derivados del cribado virtual, sugieren que moléculas tales como la ascididemina, el peyssonol B, el ácido asiático y el ácido docosahexaenoico, podrían comportarse como inhibidores débiles de proteínas involucradas en diversas patologías. Estas propiedades podrían explicar algunas de los beneficios que han sido observados para estos compuestos de origen natural, cuando son empleados para el control, mejoramiento o eliminación de diferentes enfermedades en humanos.
Agradecimientos
Los autores agradecen a Colciencias, Bogotá (Colombia), y la Universidad de Cartagena, Cartagena (Colombia) por su apoyo financiero (Proyectos: 110745921616, 2009; 110751929058, 2010); así como también al Programa de Apoyo a Grupos de Investigación de la Vice-Rectoría de Investigaciones de la Universidad de Cartagena (2009-2012), y el Programa Nacional para Estudios de Doctorados en Colombia de Colciencias, año 2011.
Bibliografía
Appleton, D. R.; Norrie Pearce, A.; Copp, B. R. 2010. Anti-tuberculosis natural products: synthesis and biological evaluation of pyridoacridine alkaloids related to ascididemin. Tetrahedron, 66(27/28):4977-4986. [ Links ]
Butler, M. S.; Buss, A. D. 2006. Natural products -The future scaffolds for novel antibiotics?. Biochem. Pharmacol. 71(7):919-929. [ Links ]
Cai, J.; Han, C.; Hu, T.; Zhang, J.; Wu, D.; Wang, F., Liu, Y.; Ding, J.; Chen, K.; Yue, J.; Shen, X.; Jiang, H. 2006. Peptide deformylase is a potential target for anti-Helicobacter pylori drugs: reverse docking, enzymatic assay, and x-ray crystallography validation. Protein Sci. 15(9):2071-2081. [ Links ]
Chen, Y. Z.; Ung, C. Y. 2002. Computational method for drug target search and application in drug discovery. J. Theor. Comput. Chem. 1(1):1-12. [ Links ]
Chomanska, U.; Krasnicki, P.; Proniewska-Skretek, E.; Mariak, Z. 2010. Acute phacomorphic glaucoma in patient with diabetes mellitus type 1-case report. Klin. Oczna. 112(7-9):240-242. [ Links ]
Cordell, G. 2000. Biodiversity and drug discovery a symbiotic relationship. Phytochemistry 55(6):463-480. [ Links ]
DeLano, W. L. The PyMOL molecular graphics system. DeLano Scientific LLC. 1998-2003. San Carlos, CA, USA. http://www.pymol.org. [ Links ]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. R.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr. J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian03. Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [ Links ]
Gad, S. C. 2007. Preclinical development handbook: ADME and biopharmaceutical properties.; Wiley-Interscience: Cary, North Carolina. DOI 10.1002/9780470249031. [ Links ]
Ganesan, A. 2008. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 12(3):306-317. [ Links ]
Gao, Z.; Li, H.; Zhang, H.; Liu, X.; Kang, L.; Luo, X.; Zhu, W.; Chen, K.; Wang, X.; Jiang, H. 2008. PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics, 9:104. [ Links ]
Guha, R.; Howard, M. T.; Hutchison, G. R.; Murray-Rust, P.; Rzepa, H.; Steinbeck, C.; Wegner J. K.; Willighagen, E. 2006. The blue obelisk -Interoperability in chemical informatics. J. Chem. Inf. Model. 46(3):991-998. [ Links ]
Harriman, J.; Deleavey, G.; Lambropoulos, A.; Deslongchamps, G. 2007. Reverse-docking study of the organocatalyzed asymmetric Strecker hydrocyanation of aldimines and ketimines. Tetrahedron, 63(52):13032-13038. [ Links ]
Harvey, A. L. 2007. Natural products as a screening resource. Curr. Opin. Chem. Biol. 11(5):480-484. [ Links ]
Harvey, A. L. 2008. Natural products in drug discovery. Drug Discov. Today. 13(19/20):894-901. [ Links ]
Jain, S.; Chincholikar, A. 2004. Pharmacophore Mapping And Drug Design. Indian J. Pharm. Sci. 66(1):11-17. [ Links ]
Jeong, C.; Bode, A. M.; Pugliese, A.; Cho,Y. Y.; Kim, H. G.; Shim, J. H.; Jeon, Y. J.; Li, H.; Jiang, H.; Dong, Z. 2009. [6]-Gingerol suppresses colon cancer growth by targeting Leukotriene A4 Hydrolase. Cancer Res. 69(13):5584-5591. [ Links ]
Ji, H.; Li, X.; Zhang, H. 2009. Natural products and drug discovery. EMBO reports. 10(3):194-200. [ Links ]
Kada, N.; Suzuki, T.; Aizawa, K.; Matsumura, T.; Ishibashi, N.; Suzuki, N.; Takeda, N.; Munemasa, Y.; Sawaki, D.; Ishikawa, T.; Nagai, R. 2007. Acyclic retinoid inhibits neointima formation through retinoic acid receptor beta-induced apoptosis. Arterioscler. Thromb. Vasc. Biol. 27(7):1535-1541. [ Links ]
Kämper, A; Apostolakis, J.; Rarey, M.; Marian , C.; Lengauer, T. 2006. Fully automated flexible docking of ligands into flexible synthetic receptors using forward and inverse docking strategies. J. Chem. Inf. Model.46(2):903-911. [ Links ]
Kaneski, C. R.; Moore, D. F.; Ries, M.; Zirzow, G. C.; Schiffmann, R. 2006. Myeloperoxidase predicts risk of vasculopathic events in hemizgygous males with Fabry disease. Neurology. 67(11):2045-2047. [ Links ]
Kansal, N.; Silakari, O.; Ravikumar, M. 2010. Three dimensional pharmacophore modelling for c-Kit receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 45(1):393-404. [ Links ]
Kapetanovic, I. M. 2008. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 171(2):165-176. [ Links ]
Kulkarni, O. P.; Sayyed, S. G.; Kantner, C.; Ryu, M.; Schnurr, M.; Sárdy, M.; Leban, J.; Jankowsky, R.; Ammendola, A.; Doblhofer, R.; Anders, H. J. 2010. 4SC-101, a novel small molecule dihydroorotate dehydrogenase inhibitor, suppresses systemic lupus erythematosus in MRL-(Fas)lpr mice. Am. J. Pathol. 176(6):2840-847. [ Links ]
Kumar, K.; Waldmann, H. 2009. Synthesis of natural product inspired compound collections. Angew. Chem. Int. Ed. Engl. 48(18):3224-42. [ Links ]
Lam, K. 2007. New aspects of natural products in drug discovery. Trends Microbiol. 15(6):279-289. [ Links ]
Lane, A. L.; Mular, L.; Drenkard , E. J.; Shearer, T. L.; Engel, S.; Fredericq,
S.; Fairchild, C. R.; Prudhomme, J.; Roch , K.; Hay, M. E.; Aalbersberg, W.; Kubanek, J. 2010. Ecological leads for natural product discovery: novel sesquiterpene hydroquinones from the red macroalga Peyssonnelia sp. Tetrahedron, 66(2):455-461. [ Links ]
Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yul, K.; Zhul, W.; Chen, K.; Shen, J.; Wang, X.; Jiang, H. 2006. TarFisDock: a web server for identifying drug targets with docking approach. Nucl. Acids Res. 34(2):219-224. [ Links ]
Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Jiang, H. 2010. PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach. Nucl. Acids Res. 38(2):609-614. [ Links ]
Maldonado-Rojas, W.; Olivero-Verbel, J.; Ortega-Zúñiga, C. 2011. Searching of protein targets for alpha lipoic acid. J. Braz. Chem. Soc. 22(12): 2250-2259. [ Links ]
Mandal, S.; Moudgil, M.; Mandal, S. K. 2009. Rational drug design. Eur. J. Pharmacol. 625(1/3):90-100. [ Links ]
Matsumoto, S.; Biggs, J.; Copp, B. R.; Holden, J. A.; Barrows, L. R. 2003. Mechanism of ascididemin-Induced cytotoxicity. Chem. Res. Toxicol. 16(2):113-122. [ Links ]
Mattingly, C. 2009. Chemical databases for environmental health and clinical research. Toxicol. Lett. 186(1):62-65. [ Links ]
McChesney, J.; Venkataraman, S.; Henri, T. 2007. Plant natural products: Back to the future or into extinction?. Phytochemistry, 68(14): 2015-2022. [ Links ]
Mukherjee, S.; Nagar, S.; Mullick, S.; Mukherjee, A.; Saha, A. 2008. Pharmacophore mapping of arylbenzothiophene derivatives for MCF cell inhibition using classical and 3D space modeling approaches. J. Mol. Graph. Model. 26(5):884-892. [ Links ]
Mullen, A.; Loscher, C. E.; Roche, H. M. 2010. Anti-inflammatory effects of EPA and DHA are dependent upon time and dose-response elements associated with LPS stimulation in THP-1-derived macrophages. J. Nutr. Biochem. 21(5):444-450. [ Links ]
Olivero-Verbel, J.; Cabarcas-Montalvo, M.; Ortega-Zúñiga, C. 2010. Theoretical targets for TCDD: A bioinformatics approach. Chemosphere, 80(10):1160-1166. [ Links ]
Pandey, J.; Sharma, A.; Tiwari, V. K.; Dube, D., Ramachandran, R.; Chaturvedi, V.; Sinha, S.K.; Mishra, N.N.; Shukla, P.K.; Tripathi, R. P. 2009. Solution phase synthesis of a library of carbapeptide analogues based on glycosylamino acid caffolds, their in silico screening and antimicrobial evaluation. J. Comb. Chem. 11(3):422-427. [ Links ]
Patil, S. P.; Maki, S.; Khedkar, S. A., Rigby, A. C.; Chan, C. 2010. Withanolide A and asiatic acid modulate multiple targets associated with amyloid-beta precursor protein processing and amyloid-beta protein clearance. J. Nat. Prod. 73(7):1196-202. [ Links ]
Ramana, K. V. ; Tammali, R. ; Srivastava, S. K. 2010. Inhibition of aldose reductase prevents growth factor-induced G1-S phase transition through the AKT/phosphoinositide 3-kinase/E2F-1 pathway in human colon cancer cells. Mol. Cancer The. 9(4):813-824. [ Links ]
RCSB PDB Protein Data Bank . www.pdb.org/pdb/home/home.do. [ Links ]
Rishton, G. M. 2008. Natural products as a robust source of new drugs and drug leads: past successes and present day issues. Am. J. Cardiol. 101(10A):44D-49D. [ Links ]
Rudolph, V.; Andrié, R. P.; Rudolph, T. K.; Friedrichs, K.; Klinke, A.; Hirsch-Hoffmann, B.; Schwoerer, A. P.; Lau, D.; Fu, X. M.; Klingel, K.; Sydow, K.; Didié, M.; Seniuk, A.; Von Leitner, E.; Szoecs, K.; Schrickel, J. W.; Treede, H.; Wenzel, U.; Lewalter, T.; Nickenig, G.; Zimmermann, W.; Meinertz, T.; Böger, R. H.; Reichenspurner, H.; Freeman, B. A.; Eschenhagen, T.; Ehmke, H.; Hazen, S. L.; Willems, S.; Baldus, S. 2010. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat. Med. 16(4):470-474. [ Links ]
Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H. J. 2008. PharmaGist: a webserver for ligand-based pharmacophore detection. Nucl. Acids Res. 36(2):223-228. [ Links ]
SYBYL molecular modeling software, Version 8.1. 2007. Tripos, St. Louis, MO, U.S.A. [ Links ]
Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. 2006. New technologies in computer-aided drug design: toward target identification and new chemical entity discovery. Drug Discov. Today Technol. 3(3):307-313. [ Links ]
Trott, O.; Olson, A. J. 2010. AutoDock Vina: improving the speed and accuracy
of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31(2):455-461. [ Links ]
Vita, J. A.; Brennan, M. L.; Gokce, N.; Mann, S. A.; Goormastic, M.; Shishehbor, M. H.; Penn, M.S.; Keaney, J. F.; Hazen, S. L. 2004. Serum Myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 110:1134-1139. [ Links ]
Wermuth, C. G.; Ganellin, C. R.; Lindberg, P.; Mitscher, L. A. 1998. Glossary of terms used in medicinal chemistry. Pure Appl. Chem. 70(5):1129-1143. [ Links ]
Wild, D.; Wiggins, G. 2006. Challenges for chemoinformatics education in drug discovery. Drug Discov. Today. 11(9/10):436-439. [ Links ]
Wilk, W.; Zimmermann, T. J.; Kaiser, M.; Waldmann, H. 2010. Principles, implementation, and application of biology-oriented synthesis (BIOS). Biol. Chem. 391(5):491-497. [ Links ]
Wolber, G.; Langer, T. 2005. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45(1):160-169. [ Links ]
Wolfe, M. S.; Selkoe, D. J. 2010. Giving Alzheimer's the old one-two. Cell. 142(2):194-196. [ Links ]
Yadav, U. C. ; Srivastava, S. K.; Ramana, K. V. 2010. Understanding the role of aldose reductase in ocular inflammation. Curr. Mol. Med. 10(6): 540-549. [ Links ]
Zameitat, E.; Freymark, G.; Dietz, C. D.; Löffler, M.; Bölker, M. 2007. Functional expression of human dihydroorotate dehydrogenase (DHODH) in pyr4 mutants of ustilago maydis allows target validation of DHODH inhibitors in vivo. Appl. Environ. Microbiol. 73(10):3371-3379. [ Links ]
Chen, Z., Wang, X., Zhu, W., Cao, X., Tong, L., Li, H., Xie, H., Xu, Y., Tan, S., Kuang, D., Ding, J., Qian, X. 2011. Acenaphtho [1,2-b] pyrrole-Based selective fibroblast growth factor receptors 1 (FGFR1) Inhibitors: design, synthesis, and biological Activity. J. Med. Chem. 54(11):3732-3745. [ Links ]
Recibido: 15 de agosto de 2012
Aceptado para publicación: 5 de septiembre de 2012.
