











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
In recent years the magneto-electric compounds have attracted scientific and technological interest in science community, because they represent very interesting potential applications in electronic devices. Into this family of compounds are the complex perovskite oxides of A2MM'O6 kind, where A is an alkaline earth and M, M' are magnetic and non-magnetic transition metal, respectively. The Sr2MnSbO6 perovskite is one compound of this type, and it has been synthetized via molten salt synthesis (MSS) method and convectional solid-state methods for example by (Cheah et al., 2006; Ivanov et al., 2009; Parada et al., 2018; Preethi-Meher et al., 2012). Experimental studies of Sr2MnSbO6 have shown that this compound presents ferroelectric and ferromagnetic behavior in the I4m, I4mm, I4mcm phases, as well as it exhibits high dielectric constant in the cubic phase Fm-3m (Antara, et al., 2011; Foster, et al., 1997; Majhi & Varma, 2007). Modifications of this compound recently have been reported by (Bazuev et al., 2021), where is studied the effect of substitutions on the phase formation, structure and magnetic properties of the Sr2MnSbO6 compound.
The theoretical study of the A2MM'O6 compounds is usually tackled using the DFT framework. Its useful accuracy and low computational cost made DFT the main tool in the prediction of properties of materials. In particular, the study of complex perovskite oxides involves effects to take in account, such as the strong correlation between electrons of the transition metals elements, which are included into the DFT formalism. For example, theoretical calculations of the Sr2MnSbO6 compound using DFT framework in its pseudopotential approach (Singh & Nordstrom, 2006) with GGA+U approximation are reported in (Persson, 2016), for the tetragonal space group I4/mmm. This phase is reported as semiconductor with band gap equal to 0.208 eV, and effective magnetic moment 4.0 piB. Moreover, DFT in the FP-LAPW version is applied in a broad range of the A2MM'O6 kind compounds by (Mir & Gupta, 2020; Bonilla et al., 2007; Cardona et al., 2008; Fajardo et al., 2012; Liu, et al., 2014; Mir & Gupta, 2021; Ortiz-Diaz et al., 2007), where are included transition metals such as Cr, Co, Fe, Ni, Mn, among others. In the above mentioned cases, those complex perovskites present the so-called half-metallic behavior. This exotic property is characterized because the material has completely polarized spin states at the Fermi level, allowing the conduction of charge with specific spin (up or down). Such behavior makes these materials candidates for applications in spintronics as sources of unique spin and high efficiency sensors.
The success of the density functional theory methods to compute properties of materials like the complex perovskite oxides, lead us to develop theoretical calculations based on density functional theory (DFT), aiming to elucidate the properties of the Sr2MnSbO6 perovskite. The theoretical calculations performed here show that all, cubic (Fm-3m) and tetragonal phases (I4m and P4/mnc), are stable phases of this compound. Our findings agree with different experimental reports of the structural parameters. In addition, in this paper we present calculations of densities of states (DOS), through which a half-metallic behavior in the cubic and tetragonal structures is demonstrated.
Calculations Details
DFT calculations employing the full potential linearized augmented plane wave method (FP-LAPW) were performed, such as is implemented in the WIEN2k code (Blaha et al., 2001), within the spin-polarized density-functional theory framework (Khon & Sham, 1965). The exchange and correlation effects were described by the generalized gradient approximation GGA of Perdew - Burke - Ernzerhof (PBE) (Perdew et al., 1996) together with the Hubbard parameter U (GGA+U). The GGA+U technique was developed during the 1990s by Anisimov and coworkers to refine DFT calculations (Anisimov et al., 1991; Anisimov et al., 1993; ). It considers a Hubbard term for the Coulomb potential that corrects mean field interactions via an on-site (atomic) parameter, U. It was chosen the value 4.0 eV for the effective parameter Ueff of the d-Mn states. The iteration for self-consistency was continued until the convergence criteria, of 1 x 10-4 Ry, was reached.
The atomic positions used as input, for each space group, are shown in the table 1. The cubic and tetragonal supercells (Fm-3m, I4m and P4/mnc spaces group, respectively) were built using the Vesta software (Momma & Izumi, 2011), as shown in the figure 1. In figure 1a, it can be seen the crystal lattice structure for the cubic phase. In the picture it
Tabla 1 Atomic positions given in crystalline units, for the Fm-3m, I4m and P/4mnc phases of the Sr2MnSbO6 perovskite.
| Space group | #225 | Fm -3m | ||
|---|---|---|---|---|
| Element | x | y | z | R MT |
| Sr | 0.2500 | 0.2500 | 0.2500 | 2,0000 |
| Sb | 0.0000 | 0.0000 | 0.0000 | 2,2000 |
| Mn | 0.5000 | 0.0000 | 0.0000 | 2,0000 |
| O | 0.2504 | 0.0000 | 0.0000 | 1,4000 |
| Space group | #87 | I4m | ||
| Element | x | y | z | R MT |
| Sr | 0.0000 | 0.5000 | 0.2500 | 1,8000 |
| Mn | 0.0000 | 0.0000 | 0.0000 | 2,0000 |
| Sb | 0.5000 | 0.0000 | 0.5000 | 2,2000 |
| O1 | 0.0000 | 0.0000 | 0.2504 | 1,4000 |
| O2 | 0.2081 | 0.2910 | 0.0000 | 1,4000 |
| Space group | #128 | P4/mnc | ||
| Element | x | y | z | R MT |
| Sr | 0.2500 | 0.2500 | 0.2500 | 1,8000 |
| Sb | 0.0000 | 0.0000 | 0.0000 | 2,0000 |
| Mn | 0.5000 | 0.0000 | 0.0000 | 2,0000 |
| O1 | 0.2504 | 0.0000 | 0.0000 | 1,4000 |
| O2 | 0.2081 | 0.7090 | 0.0000 | 1,4000 |

Figura 1 Crystal structures of the Sr2MnSbO6 perovskite. Green spheres represent Strontium atoms; Red spheres represent Oxygem atoms; The Mn 3+ ions are shown as purple spheres in the octahedra; The Sb 5+ ions are shown as yellow spheres in the octahedra. (a) Fm-3m. (b) I4m. (c) P/4mnc. (d) Top view of the Fm-3m phase. (e) Top view of the I4m phase. (f) Top view of the P/4mnc phase.
is observed the Mn3+ (purple spheres) and Sb5+ (yellow spheres) cations adopt octahedral correlations with its nearest neighbors, consisting of O2- anions. The muffin-tin radii used were 2.0, 2.2, 2.0 and 1.4 bohr for Sr, Sb, Mn and O atoms, respectively, and a 120 k-points mesh was used in a Brillouin irreducible zone, generated according to the Monkhorst-Pack scheme (Monkhorst & Pack, 1976). The separation energy between the valence and core states was taken to be equal to -6.0 Ry, and the maximum angular momentum was 1max = 10. The wave functions in the interstitial region were expanded in plane waves with cutoff Kmax = 7.0/Rmt (where Rmt is the smallest muffin-tin sphere radii inside the cell).
In the case of the tetragonal I4m lattice shown in figure 1b, there are two atomic positions for oxygen atoms, i.e., the atoms placed in top of the octahedral site (O1) and the oxygen atoms placed on the xy-plane of the octahedron (O2), as shown in table 1. The muffin-tin radii used were 1.8, 2.2, 2.0 and 1.4 bohr for Sr, Sb, Mn and O atoms, respectively, and a 144 k-points mesh was used in a Brillouin irreducible zone. The separation energy between the valence and core states was taken to be equal to -6.0 Ry, and the maximum angular momentum was 1max = 10. The wave functions in the interstitial region were expanded in plane waves with cutoff Kmax = 7.0/Rmt.
Similarly, figure 1c shows the tetragonal P/4mnc lattice. For this phase there are also two atomic positions for the oxygen atoms, axial (O1) and equatorial (O2), this is illustrated in table 1. The muffin-tin radii were 1.8, 2.0, 2.0 and 1.4 bohr for Sr, Sb, Mn and O atoms, respectively. A mesh of 140 k-points was used in a Brillouin irreducible zone. The separation energy between the valence and core states was taken to be equal to -8.0 Ry. As in the former cases, the maximum angular momentum was 1max = 10 and the wave functions in the interstitial region were expanded in plane waves with cutoff Kmax = 7.0/Rmt.
The top view of the different phases of the compound in figure 1 allows to better observe the arrangement of the octahedra in the different symmetries. In the cubic phase the octahedra are placed one on top of the other and the oxygen atoms are aligned with the axes, as shown in figure 1d. In the I4m case, the oxygen atoms are not aligned with the x-and y-axis, i.e., the octahedra present rotation around the z axis. Furthermore, the vertices of octahedra are not aligned with each other along the z axis, see figure 1e. The P/4mnc structure presents the same rotation of the octahedra around the z axis, as in the I4m phase, but in this case the vertices of octahedra are aligned along the z-axis, see figure 1f. These differences in the symmetry and arrangement of octahedra often determine the electronic behavior in compounds of this type.
Results and discussion
Structural parameters
An optimization process of the equilibrium structural parameters was performed computing the cohesive energy value of the structure for different values of the lattice constants. The energy versus volume values were fitted to the Murnaghan's equation of state (Murnaghan, 1944) with the aim to obtain the optimal structural parameters, i.e. the volume of equilibrium (V), lattice parameters of equilibrium (α and c/ α ratio), bulk modulus (B) and cohesive energy (E 0). The results of the fits are shown in table 2 which also contains some experimental values for comparison. The lattice constant obtained for the cubic phase was α = 15.16 bohr and the corresponding cohesive energy was -3.77 Ry. Also, for the tetragonal structure I4m, the lattice constant obtained was α = 10.65 bohr, the optimized ratio c/α = 1.467 and the cohesive energy -3.78 Ry. Furthermore, for the tetragonal P4/mnc phase, the lattice constant was 10.58 bohr, c/α = 1.467 and cohesive energy equal to -3.66 Ry. We highlight that the results for the lattice parameters of the cubic and I4m phases are in good agreement with the experimental values reported in (Cheah et al., 2006; Ivanov et al., 2009).
Tabla2. Structural parameters calculated for the Fm-3m, I4m and P4/mnc space groups of Sr2MnSbO6 perovskite
| Fm-3m space group | |||||
|---|---|---|---|---|---|
| Parameter | α [bohr] | V[bohr3] | B[GPa] | E 0[Ry] | |
| This work | 15.16 | 871.15 | 155.45 | -3.77 | |
| Exp. ref. (Ivanov et al., 2009) | 15.0648 | - | - | - | - |
| Exp. ref. (Cheah et al., 2006) | 15.0854 | - | - | - | - |
| I4m space group | |||||
| Parameter | α [bohr] | c/a ratio | V[bohr3] | B[GPa] | E 0[Ry] |
| This work | 10.65 | 1.43 | 866.56 | 169.59 | -3.78 |
| Exp. ref. (Ivanov et al., 2009) | 10.4161 | 1.4608 | - | - | - |
| Exp. ref. (Cheah et al., 2006) | 10.4526 | 1.4613 | - | - | - |
| P4/mnc space group | |||||
| Parameter | α [bohr] | c/a ratio | V[bohr3] | B[GPa] | E 0[Ry] |
| This work | 10.58 | 1.46 | 864.47 | 157.24 | -3.66 |
| Exp. ref. (Parada et al., 2018) | 10.688 | 1.4180 | - | - | - |
On the other hand, to our knowledge the only values reported for the parameters of the tetragonal P/4mnc phase are those experimentally obtained by (Parada et al., 2018) which are comparable to ours, as shown in the table 2. In adittion, it is noticed the negative value for the cohesive energy, which proves the stable character of this phase and consequently new evidence of its existence. The energy values let us conclude that for the tetragonal symmetry, the compound prefers the arrangement shown in figure 1e, over the one in which the octahedra are placed in layers perfectly aligned, as shown in figure 1f.
Density of States (DOS)
The electronic behaviour of the compound is determined by computing and plotting the densities of states for the up and down spin polarization. Results of this for the cubic phase are shown in figure 2. Here, it can be seen the total density of states, along with the atomic contribution and the main contributions of the atomic orbitals. In figure 2a. are shown the total density (red line) and the contribution of the different atomic species in the compound. The largest contribution to the density of states is due to O and Mn atoms whereas the Sr and Sb atoms does not contribute at least around the Fermi level (energy equal to zero). It is observed some states at the Fermi level corresponding to the spin up polarization whereas the spin down polarization does not present states at the Fermi level. This implies that the compound exhibits metallic-like behavior for the spin-up polarization, while in the spin-down polarization, the material behaves as semiconductor.

Figura 2 (a) Total density of states and atomic contribution for the cubic structure. (b) Main contri bution of the Mn-atoms. (c) Main contributions of the O-atoms.
In addition, we can notice that the states below of Fermi level are principally due to the O atoms, in both up and down spin polarization. Some spin-up states of manganese and oxygen are present at the Fermi level while minority spin electrons are negligible indicating that the majority spin electrons alone participate in the electrical conductivity, which support the charge transportation for spin-up channel.
In figure 2b and figure 2c also are shown the partial densities of states with more considerable contribution, which are mainly due to Mn - d and O - p orbitals. It is easy to notice in the DOS of the Mn - d orbitals, the splitting of crystalline field effects, which it is due the d - Mn orbitals. In a cubic transition-metal oxide crystal the transition-metal ion is in a site that has octahedral O h symmetry and the d-levels split into threefold degenerate (lower energy) t 2g states and twofold degenerate (higher energy) e g states. For the Manganese, the main contribution of t 2g states is about -5.2 eV while the e g states is about the Fermi level, for the spin up density. Furthermore, it can be observed for the down polarization that t 2g states contribute in values about 3.2 eV and the e g states are placed about the 4.2 eV. As demonstrated trough the values above the splitting of crystalline field for spin up polarization is more energetic than splitting for spin down polarization. The splitting of crystalline field is above 5.2 eV for up polarization and about 1.0 eV for down spin configuration. On the other hand, due to the magnetic characteristic of M cation, an exchange splitting takes place. It is observed as a difference between e g up and down states, and a difference between t 2g up and down states. The splitting of exchange shows approximately 4.2 eV for high energy states and 8.4 eV for low energy states. For the oxygen atoms it is found that the px + py orbitals have presence mainly in energy values less than -1.0 eV while the O-pz states are also in the Fermi level.
The densities of states for the tetragonal I4m phase are shown in figure 3. The first thing to be noticed is that there are some few states about the Fermi level, corresponding to the spin up polarization. As in the cubic case, the O and Mn atoms provide states about the Fermi level while the absence of Sr and Sb states persist (see figure 3a). The manganese provides occupied states, mainly in the up spin polarization, which can be seen in figure 3b. At energies below the Fermi level, the main contribution to the DOS is due to the oxygen atoms, as shown in figure 3c and figure 3d.
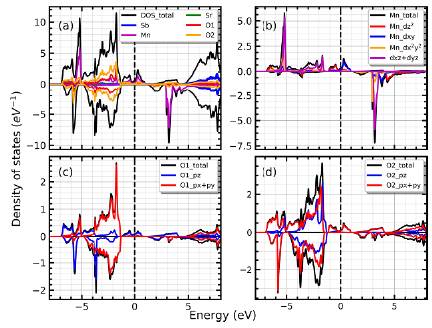
Figura 3 (a) Total density of states and atomic contribution for the tetragonal I4m phase. (b) Main contribution of the Mn-atoms. (c) DOS of the O1-atoms placed at the top of octahedra. (d) Dos cor responding to the O2-atoms placed in the x-y plane of the octahedra.
In figure 3b the DOS for each d - Mn orbitals also are shown. In this case the orbitals are not splitted in two groups as in the cubic case, due the tetragonal distortion and the oxygen-octahedron rotation. The splitting of crystal field is affected by the atomic spacing increase in the z-direction due the change to tetragonal symmetry and the arrangement of the octahedra. The so called, Jahn-Teller effect, describes this type of distortions which depends on both the strength and the symmetry of the crystal field. In this case, the bond length between the metal ion and the O1 atoms is slightly longer than the bond lengths with the O2 atoms. Also, the vertices of the octahedron are not aligned with the x- and y-axis, but rather it is rotated with respect to the z axis, as can be seen in figure 1e. The above destabilize the orbitals with components in the x-y plane (higher energy) and the orbitals with components along the x- and y-axis are stabilized (lower energy). The combination of the two distortions is responsible for the contributions of states near the Fermi level due the d z2 and the d xy orbitals, whereas the dx2+y2 and d xz+yz orbitals contribute at energies below the Fermi level.
The DOS for the O1 and O2 atoms are shown in figure 3c and figure 3d, respectively. In figure 3c it can be seen the DOS of O1 atoms, recalling that these atoms are placed at top of the octahedral site. The O1-px + py states are scattered on energy values from -7.0 to -1.5 eV, in both up and down polarizations, whereas the O1 -pz also are present about the Fermi level, only for spin up channel. Meanwhile, in figure 3d one sees that O2-pz orbitals are scattered along the energy axis from -7.0 to -1.5 eV, whereas the O2-px + py orbitals also provide states about the Fermi level. So, the Mn-d z2 , Mn-d xy , O1-Pz and O2-px+py states support the charge transport with spin up polarization. The spin down channel still behaves like a semiconductor, as in the previous case.
Finally, let us discuss the densities of states computed for the P/4mnc phase, which are shown in the figure 4. It is easy to notice that the DOS for the P/4mnc are very similar to those shown in figure 3, for the former case. In figure 4a, it is observed that near the Fermi level most of the states correspond to Mn, O1 and O2 atoms. Partial densities of states showing various orbital contributions for these atoms are presented in figure 4b,4c and 4d, respectively. Here it is possible to see that the interaction between Mn-d and O-p states produces some states about the Fermi level, appearing the metallic character in the spin up channel, while the spin down channel exhibits semiconductor behaviour. The similarity of the densities of states with the I4m phase allows us to infer that the P/4mnc phase, besides to implying the appearance of more states at the Fermi level, does not have a significant effect on the overall electronic behavior.
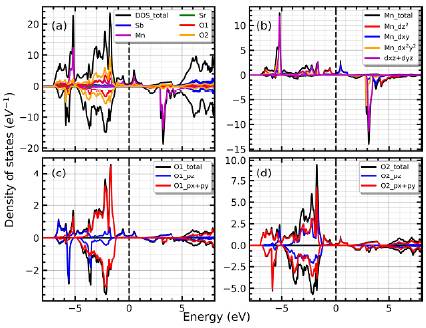
Figura 4 (a) Total density of states and atomic contribution for the tetragonal P/4mnc phase. (b) Main contribution of the Mn-atoms. (c) DOS of the O1-atoms placed at the top of octahedra. (d) Dos corresponding to the O2-atoms placed in the x-y plane of the octahedra.
In all the cases studied the DOS is asymmetric with respect to the spin polarization. This asymmetrical behavior permits to obtain an effective magnetic moment. The value found was 4.0 µB per unit cell in both Fm-3m and I4m phases, and 8.0 µB for the P4/mnc phase.
In addition, based on the asymmetry of all the projections on the Mn orbitals, it was concluded that the magnetic moment is principally due the Mn-d orbitals contribution.
Conclusions
Theoretical calculations of density functional theory have been utilized in our study of the structural and electronic properties of the perovskite, Sr2MnSbO6. Our results are in agreement with experimental reports of structural parameters, as well as the cohesive energy and the bulk modulus are reported. Also, it is reported the p/4mnc phase whose main difference from the I4m is the arrangement of the octahedra, as shown in figure 1e and figure 1f, and as a consequence the material prefers the I4m arrangement since it has a more negative energy value (more stable) than the P/4mnc phase. The densities of states reveal a half-metallic behavior in the Sr2MnSbO6 compound, which is supported by the presence of Mn-d and O-p states about the Fermi level. Also, it is observed the asymmetric shape of the DOS with respect to the spin polarization, which implies the existence of effective magnetic moment.

