











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Fullerenes, and especially [60]fullerene, have attracted great interest in the scientific community since their discovery by R. R. Curl, H. W. Kroto y R. E. Smalley in 1985 (Kroto et al, 1985) and the development of large-scale methods to obtain them (Krätschmer et al., 1990). These carbon allotropes exhibit very interesting photophysical and electrochemical properties, including low reorganization energy, the ability to reversibly accept up to six electrons, easily excited by low-energy light, and high electron affinities, among others (Acquah et al, 2017). These properties have motivated synthetic chemists and materials scientists to design and prepare new fullerene derivatives that can be potentially used in different fields from medicine to molecular electronics (Taylor et al, 1993; Hirsch et al, 2005; Bakry, et al, 2007; Page et al, 2014).
On the other hand, hydrazones are a family of compounds incorporating in their structure the R1-C=N-NH-R2 moiety. They can undergo configurational dynamics by photochemical and thermal E/Z isomerization and form complexes with transition metals (Chaur et al, 2011). Besides, the relative ease of the synthetic routes and the capacity to change its structure with different substituents are properties that have been explored in areas such as supramolecular chemistry, metal-organic structured materials, dynamic combinatorial, and cation and anion detection, among others (Galindo-Betancourth et al., 2022).
Previously our research group reported the synthesis of a pyrrolidino[60]fullerene derivative with a hydrazone group in its structure (Romero et al., 2017). However, the low solubility of this molecule in common organic solvents prevented the study of the photophysical and electrochemical properties. Hence, here we report the synthesis of a new pyrrolidino[60]fullerene derivative, highly soluble in different organic solvents, that exhibits the photophysical and electrochemical properties of [60] fullerene and the previously reported characteristics of the hydrazones. Given its high solubility in common organic solvents, the fulleropyrrolidine, reported herein for the first time, will allow the study of coordination properties for the self-assembly of components in supramolecular systems with a prospective in molecular electronics.
Experimental section
All starting reagents for the synthesis of the reported compounds were purchased from Sigma-Aldrich, Merck, and Alfa Aesar (USA) and were used without further purification. Reactions were monitored by TLC using silica gel 60F 254 plates with a 0.2 mm thickness (Merck) and manually revealed with a UV lamp Spectroline Series E with two wavelengths (254 and 365). 1H, 13C NMR spectra were taken in a Bruker UltraShield 400 MHz instrument using as solvents DMSO-d6 and CDCl3 according to the case. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was conducted in positive mode. Ultraviolet spectra were taken in a UV-Vis Jasco V-730 spectrophotometer. The electrochemical study was registered in voltammograms in a potentiostat (model PGSTAT302N, Methrom Autolab).
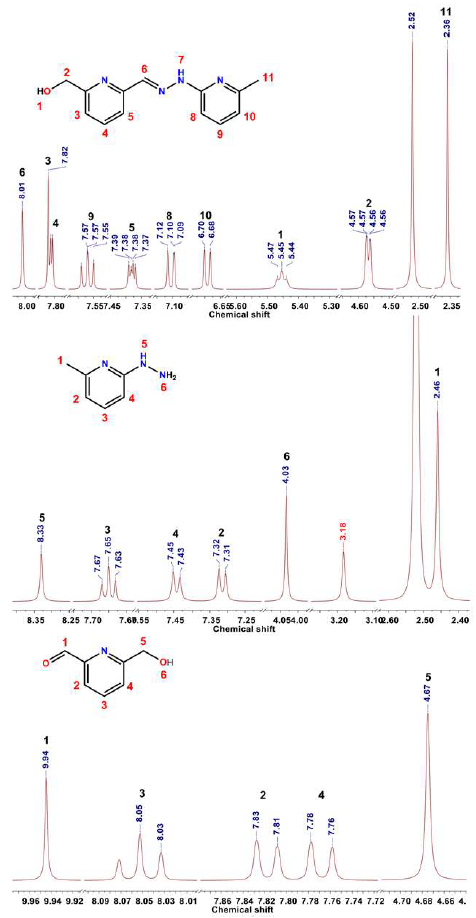
6-(hydroxymethyl)-2-pyridinecarboxaldehyde (2). MnO2 (1.26 g, 14.5 mmol, 5 equiv) was added to a solution of 2,6-dihydroxymethylpyridine 1 (0.40 g, 2.9 mmol, 1 equiv.) in CHCl3 (10 mL) and stirred for 6 h at 60 °C. The solution was then filtrated discarding the solid, and the solvent was evaporated under reduced pressure. The resulting compound was purified using column chromatography with CHCl3 obtaining MeOH 5% and yellow oil. Yield: 0.18 g, 45%. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 9.94 (s, 1H), 8.05 (t, J = 7.7 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 4.67 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ/ppm: 194.1, 163.1, 151.7, 138.7, 125.5, 120.8, 64.2.
2-hydrazinyl-6-methylpyridine (4). A solution of 2-bromo-6-methylpyridine 3 (0.30 g, 1.75 mmol, 1 equiv.) in hydrazine monohydrate (0.85 mL, 17.5 mmol, 10 equiv.) was taken to reflux in an oil bath for 24 h under inert atmosphere. Later, the hydrazine monohydrate excess was removed under reduced pressure. The resulting solution was treated with potassium carbonate (K2CO3) under stirring; the liquid phase was extracted with CHCl3, and after drying the different portions, compound 4 was obtained as a white solid (0.14 g) with a 65% yield. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 8.33 (s, 1H), 7.65 (t, J = 7.7 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 4.01 (s, 1H), 3.18 (s, 1H), 2.46 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ/ppm: 140.2, 125.5, 123.2, 79.7, 63.6, 24.0.
(E)-6-hydroxymethyl-2-pyridinecarboxaldehyde-2'-pyridyl-6'-methylhydrazone (5). A solution 2 (0.10 g, 0.7 mmol, 1 equiv.) in ethanol (5 mL) was added to a solution of 4 (0.09 g, 0.7 mmol, 1 equiv.) in ethanol (1 ml). The resulting yellow precipitate was decantated and recrystallized from ethanol to obtain the product as a light-yellow solid (0.15 g) with an 85% yield. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 11.11 (s,1H), 8.01 (s, 1H), 7.81 (d, J = 5.6 Hz, 2H), 7.57 (t, J = 7.8 Hz, 1H), 7.38 (dd, J = 5.6, 3.1 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.69 (d, J = 7.3 Hz, 1H), 5.45 (t, J = 5.9 Hz, 1H), 4.57 (d, J = 4.8 Hz, 2H), 2.36 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ/ppm: 162.0, 156.7, 153.8, 139.5, 138.9, 137.4, 119.6, 117.4, 115.1, 103.9, 64.6, 24.1.
(E)-6-carboxaldehyde-2-pyridinecarboxaldehyde-2'-pyridyl-6'-methylhydrazone (6). MnO2 (0.04 g, 0.4 mmol, 2 equiv.) was added to a solution of 5 (0.05 g, 0.2 mmol, 1 equiv.) in CHCl3 (20 mL) and stirred for 24 h at room temperature. Then, more MnO2 (0.04 g, 0.4 mmol, 2 equiv.) was added and the reaction was finished 24 h later. The resulting compound was purified using column chromatography with CHCl3: MeOH 5%, obtaining an orange solid (0.02 g) with a 50% yield. 1H NMR (400 MHz, DMSO-d6) δ/ppm: 10.13 (s, 1H), 10.07 (s, 1H), 8.11 (d, J = 4.3 Hz, 1H), 8.08 (s, 1H), 7.89 - 7.85 (m, 2H), 7.62 - 7.53 (m, 1H), 6.73 (d, J = 6.7 Hz, 1H), 6.68 (d, J = 7.4 Hz, 1H), 2.43 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ/ppm: 193.2, 154.9, 152.4, 138.4, 138.3, 137.2, 123.8, 121.0, 116.8, 107.7, 29.7.
Pyrrolidino[60]fullerene (7). A solution of C60 (0.20 g, 0.28 mmol, 1 equiv.), 6 (0.26 g, 1.12 mmol, 4 equiv.), and V-octyl glycine (0.32 g, 1.7 mmol, 6 equiv.) in 65 mL of dry toluene was heated under reflux for 30 min under an inert atmosphere of argon and coupled to a Dean-Stark trap. After one hour, the solvent was removed under reduced pressure; the resulting dark solid was purified by column chromatography on silica gel eluting with CS2 to remove the unreacted C60 and, then, using toluene: ethyl acetate 10% to give 7 as a dark brown solid (0.1 g) with 35 % yield. 1H NMR (400 MHz, CDCl3) δ/ppm: 8.28 (dd, J = 8.0, 1.2 Hz, 1H), 8.02 (t, J = 7.8 Hz, 1H), 7.94 (dd, J = 7.6, 1.2 Hz, 1H), 7.80 (d, J = 2.3 Hz, 1H), 7.75 (d, J = 9.2 Hz, 1H), 7.25 (dd, J = 9.2, 6.6 Hz, 1H), 6.60 (d, J = 6.7 Hz, 1H), 5.42 (s, 1H), 5.15 (d, J = 9.4 Hz, 1H), 4.20 (d, J = 9.4 Hz, 1H), 3.08 (dt, J = 11.8, 8.2 Hz, 1H), 2.69 (dd, J = 8.1, 4.0 Hz, 1H), 2.31 (s, 3H), 1.37 (m, 5H), 1.30 - 1.23 (m, 8H), 0.99 - 0.88 (m, 3H). MALDI-TOF MS: m/z exp. 1086.109 (C82H31N5 cal. 1085.270). Elemental analysis for C82H31N5. Calculated: C 90.68%, H 2.88%, N 6.45%. Found: C 90.72%, H 2.84%, N 6.42%.
Results and discussion
Synthesis and purification of fullerene derivative (7)
Scheme 1 shows the five-step synthetic route to obtain the pyrrolidino[60]fullerene 7. The first step implied the oxidation of the hydroxymethyl-pyridine 1 using an oxidizing agent (MnO2), leading to the formation of the aldehyde derivative 2 with a 45% yield. On the other side, the bromide compound 3 was subjected to a nucleophilic aromatic substitution reaction using hydrazine monohydrate as solvent under reflux for 24 h. The hydrazine excess was removed under reduced pressure, and the product obtained was re-dissolved in chloroform. The resulting solution was treated with K2CO3 under constant stirring. The liquid phase was extracted with CHCl3 and the extract was taken to dryness obtaining compound 4 as a white solid with a 65% yield. This compound showed to be sensitive to air and humidity and, therefore, used as soon as possible in the next reaction.

Scheme 1 Synthetic route to obtain the Mleropyrrolidine (7). (a) MnO2 CHCl3, r.t, 45%; (b) N2H4 monohydrate, reflux, 65%; (c) ethanol, reflux, 85%; (d) MnO2 CHCl3, ' r.t, 45%; (e) C60, W-Octyl glycine, toluene, reflux, 35%
After obtaining compound 4, we dissolved it in ethanol and added it dropwise to an ethanolic solution of 2. Almost immediately, a yellow solid precipitated, which was then recrystallized in cold ethanol yielding compound 5 with an 85% yield as a beige solid. Subsequently, hydrazone 5 was oxidized using MnO2 as an oxidizing agent leading to the formation of the hydrazone-aldehyde derivative 6 with a 50% yield. Compounds 2,4 and 5 were characterized using 'H NMR spectroscopy and revealed all the right number of characteristic signals assigned in figure 1.
Following the synthetic procedure, the next step involved the addition of hydrazone 6 to C60 via a Prato reaction, as previously reported (Maggini et al., 1993). This functionalization of C60 involves a 1,3-dipolar addition of the azomethine ylide generated in situ by the decarboxylation of the immonium salt derived from the condensation of W-Octyl glycine with aldehyde 6. For this purpose, C60, aldehyde 6, and W-Octyl glycine in toluene were taken to reflux under an inert atmosphere. Due to the symmetry of [60] fullerene, it is common to obtain multi-addition products. To avoid this, the reaction was constantly monitored by TLC, and once these sub-products were observed, it was stopped. The characteristic purple color of the [60]fullerene solution changed to a dark brown, a common observation in Prato reactions. Later, the resulting products were purified using column chromatography. Unreacted C60 was recovered using CS2 and toluene: ethyl acetate 10% was used to obtain 7 as a dark brown solid with a 35% yield. This compound is soluble in chloroform, dichloromethane, tetrahydrofuran, ortho-dichlorobenzene, and toluene, among others, useful in photophysical and electrochemical studies.
Fulleropyrrolidine 7 was characterized by 'H NMR spectroscopy (Figure 2). There were some notable changes in the aldehyde 6 spectrum, among them, the disappearance of the proton corresponding to the aldehyde group at 10.07 ppm and the presence at 7.82 ppm of the imine group proton, a well-resolved aromatic region between 7.20 and 8.02 ppm. Finally, it is important to mention the presence of the the pyrrolidine ring protons as two doublets at 5.15 and 4.21 ppm and a singlet at 5.42 ppm.
To correctly assign the peaks, we carried out COSY 1H-1H NMR on compound 7 (Figures 3, 4). First, we focused on the aliphatic protons in the lower frequency part of the spectrum. In Figure 3, the aliphatic chain methylene protons (H-12 in Figure 2) are labeled in red and blue circles. They appear as two double quartets at 3.10 (J ab = 8.2 Hz, J bb , = 11.8 Hz) and 2.70 ppm (Jab = 8.1 Hz, J bb , = 4.0 Hz). This multiplicity responds to the coupling with the geminal proton whose coupling constant is J bb „ and with the methylene protons labeled in a green circle in Figure 3 (H-13 in Figure 2). The correlation of the methylene protons is highlighted with a blue dashed square in the COSY spectrum (Figure 3).


On the other hand, the two doublets at 5.15 ppm (J c , = 9.4 Hz) and 4.21 ppm (J c , = 9.4 Hz) corresponded to the diastereotopic protons, labeled with purple and brown circles in Figure 3 (H-1 in Figure 2) on carbon 5 of the pyrrole ring, while the singlet at 5.42 ppm to the proton on carbon 2 of pyrrole, labeled with a magenta star. The aromatic protons were assigned based on the coupling constants from the 1H NMR and their COSY correlations (Figure 4). Although the 13C NMR spectrum could not be obtained due to the low amount of compound 7, valuable information was still obtained from the NMR data collected. The aromatic region was well represented and the lower resolution signals, characteristic of the fullerenes, were also observed. We also performed an elemental analysis that had an excellent agreement with the calculated data. These results demonstrated the success of the synthetic approach and provided important information for further characterization of compound 7.
On the other hand, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was carried out for compound 7 (Figure 5). The mass spectrum was also compared to the simulated isotopic distribution pattern finding a good correlation.


Photophysical properties of the fulleropyrrolidine 7
The photophysical properties of the fullerene derivative were studied by UV-Vis spectroscopy in toluene and compared to those of the hydrazone 6 and pristine [60] fullerene (Figure 6). Compound 7 exhibits absorption bands centered around 326 and 431 nm, which are common for fullerene adducts and are attributed to the fullerene framework π-π * transitions, and to singlet-singlet-allowed transitions, respectively. It is worth noting the hypsochromic shift and lower extinction coefficient due to the saturation of a double bond on the C60 leading to a partial loss of conjugation in the n-system and its symmetry. Finally, the band centered around 700 nm can be attributed to a charge transference process.

Electrochemical studies
Cyclic and square wave voltammetry studies were carried out for 6, 7, and [60]fullerene in 0.1 M solutions of tetra-n-butylammonium hexafluorophosphate (NBu4PF6) used as supporting electrolyte in THF. A 3 mm glassy carbon electrode served as a working electrode, a silver wire as a pseudo-reference electrode, and a wire of platinum as a counter electrode. Ferrocene was added at the end of the experiment taking its oxidation potential as an internal reference. Figure 7 shows the cyclic voltammetry (CV) and Osteryoung square wave (OSWV) voltammograms. The peak potentials, determined by OSWV, are reported in table 1.

Figure 7 Cyclic voltammetry and square wave voltammetry of 7 and [60]fullerene in 0.1 M solutions of NBu4PF6 in THF. Scan rate: 100 mV s-1
Table 1 Potential reductions and oxidations of 6, [60]fullerene, and 7 (vs. Fc/Fc+). Reported in volts vs ferrocene in 0.1 M NBu4PF6 and THF at 25°C
| Compound | E1 Red/V | E2 Red/V | E3 Red/V | E4 Red/V |
| 6 | -1.77 | -2.29 | -2.63 | -2.35 |
| C60 | -1.04 | -1.44 | -1.89 | |
| 7 | -1.11 | -1.70 | -2.28 |
In the OSWV voltammogram, fulleropyrrolidine 7 exhibited three reduction peaks from -1.11 V to -2.28 V that shifted towards cathodic potentials compared to [60]fullerene. Its first reduction shifted about 70 mV because 7 undergoes a loss of conjugation due to the saturation of a double [6,6] bond in the fullerene core, as previously stated. The second reduction peak, around -1.70 V, seemed to overlap with another peak of the same current intensity due to the analyte adsorption on the working electrode, as determined by CV (vide infra), which implies diffusion-controlled processes. The same phenomenon was observed for the second reduction of [60]fullerene.
To validate the diffusion-controlled process as the cause of this phenomenon, we conducted experiments using different scan rates for the first, second, and third reduction peaks of 7 (Figure 8) considering that, according to the Randles-Sevcik equation (Bard & Faulkner, 2001), a linear relationship between the peak currents of each event and the square root of the scan rate indicates a diffusion-controlled process.

From the slope of plots of the current peak (i p ) versus the square root of the scan rate, we determined that there was a linear relation and, thus, the reduction peaks in all three cathodic events were mass-transfer limited processes. In general, the loss of conjugation cathodically shifts the reduction potentials of compound 7 when compared with [60] fullerene; likewise, the reduction events lack electrochemical reversibility.
Finally, the methodology we present here may serve as a starting point for the synthesis of new metal-transition coordination compounds since the hydrazone pyridine-hydrazonepyridine framework, introduced over the fullerene, can coordinate different metal ions (Galindo-Betancourth et al., 2022).
Conclusions
A new pyrrolidine [60]fullerene 7 was successfully synthesized with a moderate yield via 1,3-dipolar cycloaddition or Prato reaction to [60]fullerene. This adduct is highly soluble in common organic solvents, which makes it an attractive option for various applications in materials science research. The electrochemical properties were analyzed using CV and OSWV voltammetry which showed cathodic shifts on the reduction peaks for 7 when compared to [60]fullerene. The terpyridine-like moiety on the fullerene surface of compound 7 opens up new possibilities for the coordination of transition metal ions.

