











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
La ferritina es una proteína intracelular altamente conservada desde los procariotas hasta los eucariotas cuya función principal es almacenar aproximadamente 4.500 átomos de Fe3+ (ac); está compuesta por 24 subunidades que se autoensamblan para formar una nanojaula de forma esférica (Figura 1A) con 14 canales: seis hidrofílicos con simetría triple, que permiten la entrada del hierro, y ocho hidrofóbicos con simetría cuádruple, los cuales proporcionan una vía de salida del hierro almacenado dentro de la proteína (Iqbal et al., 2021).
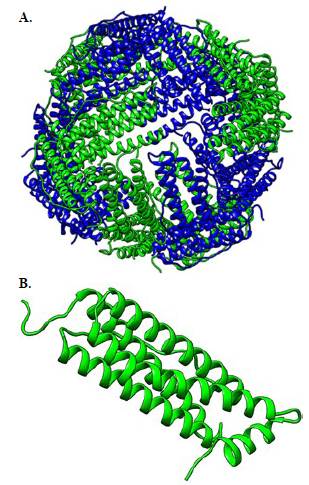
Figura 1 Modelo tridimensional de la proteína ferritina en formato de cintas: A. Estructura esférica de la proteína ferritina, conformada por 24 subunidades: en azul se representan las cadenas pesadas (FTH1) y en verde, las cadenas ligeras (FTL). B. Subunidad individual de la ferritina de cadena ligera (FTL); se resaltan los extremos N-terminal y C-terminal, que corresponden a regiones claves en la estructura y función de la proteína.
Las subunidades de la ferritina están constituidas por un 10 a 20 % de cadenas pesadas (FTH1) y un 80 a 90 % de cadenas ligeras (FTL), en las que la transcripción de los genes FTL y FTH1, que codifican las subunidades L y H de la proteína, está modulada por citocinas proinflamatorias y factores de crecimiento a través de mecanismos de regulación transcripcional (Torti & Torti, 2002). Por otro lado, la cadena ligera de la ferritina, o FTL (Figura 1B), está compuesta por 175 aminoácidos y posee variantes con cambio de sentido asociadas a ciertas enfermedades. Tal es el caso de la mutación A96T, hallada en pacientes con neuroferritinopatía (Gómez et al., 2024), y la mutación T30I, presente en pacientes aquejados por hiperferritinemia con cataratas (Carrillo et al., 2015).
La hiperferritinemia con cataratas, también conocida como síndrome de cataratas por hiperferritinemia hereditaria (Hyperferritinemia-cataract syndrome, HHCS, Online Mendelian Inheritance in Man®, OMIM #600886), es una rara enfermedad genética autosómica dominante, con una prevalencia estimada de 1/200.000 (Eris et al., 2023), que se caracteriza por la presencia de cataratas de inicio tardío y un aumento de la concentración de L-ferritina (Lachlan et al., 2004). En otras palabras, el síndrome de hiperferritinemia con cataratas es un trastorno caracterizado por un exceso de ferritina en la sangre y los tejidos del cuerpo (Yang et al., 2021).
El HHCS fue descrito por primera vez por Beaumont et al. (1995) y Girelli et al. (1995) a partir de investigaciones en varias familias. Bonneau et al. describieron tres generaciones de una familia en Francia cuyos miembros presentaban deterioro de la agudeza visual desde temprana edad, con depósitos en todas las capas de ambos cristales, un nivel elevado de ferritina circulante y ausencia de otras anomalías hematológicas o bioquímicas. Por su parte, Girelli et al. (1995) describieron dos familias del norte de Italia, una de ellas con evidencia de cataratas nucleares congénitas y otra, de cataratas bilaterales de progresión lenta con aumento en los niveles de ferritina sérica; sus análisis de hierro y los parámetros hematológicos eran normales (Craig et al., 2003a; Nos et al., 2021). Sin embargo, la información detallada sobre el fenotipo y las características de esta enfermedad apareció después, a partir del 2001 (Kaarniranta et al., 2023).
El HHCS está asociado con mutaciones en los elementos sensibles al hierro (IRE), estructuras conservadas en forma de tallo-bucle localizadas en las regiones no traducidas (UTR) 5' o 3' de los ARNm de los genes regulados por hierro, como el gen FTL (Neves et al., 2019). Estos elementos controlan la regulación postranscripcional del equilibrio u homeostasis intracelular del hierro (Khan et al., 2023). Hasta la fecha se han descrito al menos 36 mutaciones puntuales, nueve deleciones y dos inserciones en esta región (Nos et al., 2021). La mayoría de estas variantes suele ser heterocigota, lo que se explica por ser el HHCS una enfermedad de herencia autosómica dominante. Por otra parte, Kannengiesser et al. (2009) reportaron una mutación con cambio de sentido en pacientes heterocigotos en la posición c.89C>T (NM 000146.3) del gen FTL, la cual resulta en el cambio de aminoácido p. Thr30Ile (Kannengiesser et al., 2009) y explica la aparición de la enfermedad por la presencia de mutaciones puntuales en el IRE de la ferritina de cadena ligera, con la consecuente acumulación anormal de ferritina en la tejidos y suero sin sobrecarga de hierro (Kannengiesser et al., 2009).
Como ya se mencionó, entre las manifestaciones clínicas más destacadas del HHCS se encuentra la ferritina sérica elevada, mayor a 300 ng/mL en hombres y 200 ng/mL en mujeres (Elsayed et al., 2016), sin sobrecarga de hierro, así como la formación de cataratas en niños. Estas tienen diferentes fenotipos, siendo el más común la catarata bilateral con manchas blancas axiales y periféricas con agregados cristalinos (Chang-Godinich et al., 2001). Generalmente, la aparición de cataratas y síntomas ocurre en la infancia o la adolescencia, con curso progresivo, aunque en ocasiones se detectan accidentalmente en los exámenes oculares de rutina (Craig et al., 2003b). También se han descrito casos en los que los primeros síntomas se evidencian en la adultez (Kaarniranta et al., 2023).
Aunque la hiperferritinemia en este trastorno no suele causar ningún problema de salud aparte de las cataratas, los niveles elevados de ferritina en la sangre pueden confundirse con otras condiciones, como la enfermedad hepática aguda o crónica, el consumo de alcohol, las afecciones inflamatorias sistémicas, las lesiones y las neoplasias malignas (Sandnes et al., 2021). Dado que la HHCS es una enfermedad rara, suele diagnosticarse erróneamente como hemocromatosis hereditaria (HH) debido a la detección de hiperferritinemia, un signo bioquímico común de ambas enfermedades. Esto puede llevar a la administración de tratamientos equivocados, como las venoclisis o las flebotomías, que pueden ser potencialmente perjudiciales para los pacientes con HHCS, ya que terminan provocando anemia ferropénica (Giansily et al., 2013). En este contexto, el objetivo de nuestro estudio fue describir los mecanismos que pueden generar cataratas a partir de modificaciones postraduccionales de la proteína ferritina de cadena ligera.
Materiales y métodos
Modelo de caracterización en bases de datos
Se usaron las bases de datos del National Center for Biotechnology Information (NCBI) utilizando el código NP 000137.2, la Uniprot con el código P02792, el OMIM con el # 600886 y la Orphanet con el 163, para caracterizar la proteína ferritina de cadena ligera de tipo salvaje (FTL-wt) en Homo sapiens y la mutación con cambio de sentido T30I y recoger la información de base sobre esta mutación, incluida la definición de la enfermedad, el año en la que fue descrita por primera vez, su epidemiología y etiología, etc., lo que permitió una orientación clave sobre la variante.
Análisis bioinformático de las variantes proteicas
Se evaluaron diferentes características de la proteína de tipo salvaje (Wild Type) y la variante T30I, incluyendo el análisis de glicosilación, en el programa NetNGlyc 1.0 en el que, a partir de redes neuronales, se predicen los sitios de N-glicosilación. A partir de las secuencias primarias de aminoácidos de cualquier proteína, el software NetNGlyc 1.0 permite evaluar la adición de un carbohidrato a la cadena lateral de un residuo de asparagina (Asn). Esta modificación puede afectar el funcionamiento de la proteína, influyendo así en la resistencia a la proteólisis, la estabilidad de la estructura, su vida útil en la circulación y su inmunogenicidad (Gupta & Brunak, 2002).
El programa NetOGlyc 4.0 se utilizó para el análisis de la O-glicosilación a partir de las predicciones de redes neuronales de los sitios de O-glicosilación de la GalNAc de tipo mucina en proteínas de mamíferos ligada a residuos de serina y treonina. En este programa los sitios con puntuaciones > 0,5 se predicen como glicosilados y se catalogan como positivos (Steentoft et al., 2013).
Predicción de la fosforilación y activación de las rutas bioquímicas
El servidor NETPHOS 3.1 se usó para predecir sitios de fosforilación en residuos de serina (S), treonina (T) o tirosinas (Y) susceptibles de fosforilación por diversas quinasas, como la PKC, la PKA, la DNA-PK, la cdc2 y la CKI, entre otras. Cuando el software identifica un potencial sitio de fosforilación, pero no logra vincularlo con seguridad a una quinasa específica de su base de datos, etiqueta ese sitio como "unsp", indicando que se trata de una predicción no específica (Blom et al., 1999). La puntuación de predicción posee un rango entre 0 y 1, en el que las puntuaciones mayores a 0,500 indican predicciones positivas para sitios de fosforilación (Blom et al., 1999, 2004).
Con base en los resultados obtenidos, se observó un cambio significativo en la ruta metabólica mediada por la quinasa CKI en comparación con la proteína FTL-wt. Por ello, se procedió a analizar esta ruta mediante el programa Reactome Pathway Database, una base de datos biológicos de la infraestructura Elixir y GCBR, la cual proporciona información molecular seleccionada manualmente sobre una amplia gama de procesos biológicos normales y otros relacionados con enfermedades (Fabregat et al., 2018; Milacic et al., 2024).
Resultados
Caracterización bioinformática de la proteína FTL-wt y la mutación con cambio de sentido T30I
La proteína FTL-wt está compuesta por 175 aminoácidos en su secuencia primaria, y por diez sitios de fosforilación. Entre ellos se destaca la serina 33 (S-33), el cual es reconocido por la quinasa CKI (Figura 2A). En contraste, la mutación con cambio de sentido T30I posee la misma cantidad de aminoácidos, pero solo tiene nueve sitios de fosforilación debido a la ausencia del sitio S-33 fosforilado por la quinasa CKI (Figura 2B).
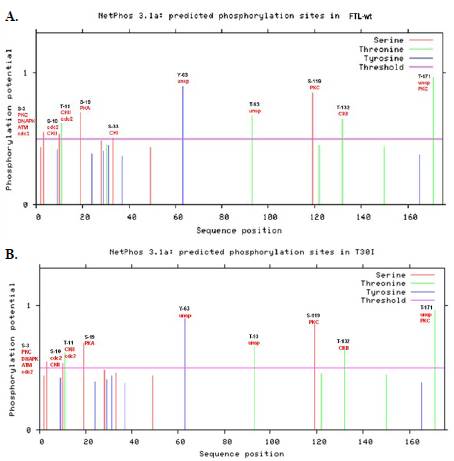
Figura 2 Caracterización bioinformática de los sitios de fosforilación hallados mediante el servidor NETPHOS 3.1. Los residuos de serrina se muestran en rojo, los de treonina en verde y los de tirosina en azul; la línea horizontal fucsia corresponde al umbral de predicción (threshold). El eje "x" representa la posición de los aminoácidos en la secuencia y el eje "y" indica el potencial de fosforilación (0 a 1). A. Predicción de sitios de fosforilación para la proteína FTL-wt, donde se destaca el residuo de serina 33 (S-33) como sitio de fosforilación reconocido por la quinasa CKI. B. Predicción de sitios de fosforilación para la mutación con cambio de sentido T30I, en la que se observa ausencia del sitio de fosforilación S-33 por la quinasa CKI.
El análisis de fosforilación consideró la participación de 17 quinasas y se centró en las fosforilaciones plausibles para los residuos de treonina, serina y tirosina, dada la presencia en sus cadenas laterales del grupo funcional hidroxilo, que favorece la fosforilación y regula la activación de rutas de señalización. Este hallazgo indicó que la variante elimina un punto crítico de regulación postraduccional de la proteína.
En cuanto al análisis de las N-glicosilaciones, se observó que ambas proteínas presentan resultados similares en la posición 8, una región polar de la secuencia de aminoácidos correspondiente al sitio de glicosilación (NYST). La probabilidad de modificación de la FTL-wt es del 0,7381 (Figura 3A), mientras que en la mutación T30I este valor aumenta levemente a 0,7382 (Figura 3B), lo que no representa un cambio significativo a nivel bioinformático. Tampoco se identificaron sitios de O-glicosilación en ninguna de las dos proteínas, resultados que se evidencian en la Tabla 1.
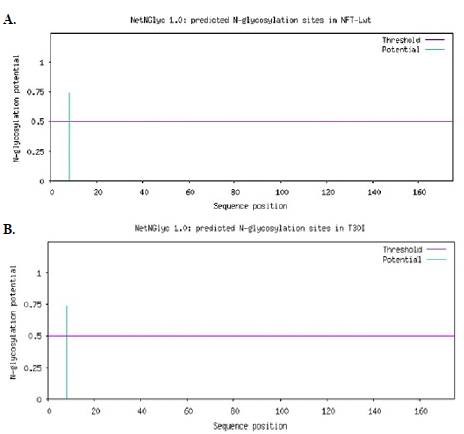
Figura 3 Caracterización bioinformática de los sitios de N-glicosilación mediante el servidor NetNGlyc 1.0. La línea horizontal púrpura corresponde al umbral de predicción (threshold). El eje "x" representa la posición de los aminoácidos en la secuencia y el eje "y" indica el potencial de N-glicosilación. A. Modificación postraduccional de N-glicosilación de la proteína FTL-wt. B. Mutación postraduccional de N-glicosilación de la mutación T30I.
Activación de rutas bioquímicas
El análisis de la mutación T30I mostró la pérdida de señal de fosforilación mediada por la quinasa CKI en comparación con la proteína FTL-wt. Este resultado motivó la evaluación de las posibles consecuencias a nivel de rutas de señalización en Homo sapiens haciendo uso de la base de datos Reactome (Figura 4A). El análisis bioinformático permitió detectar la alteración de algunas vías, entre ellas la vía de señalización por WNT (Figura 4B).
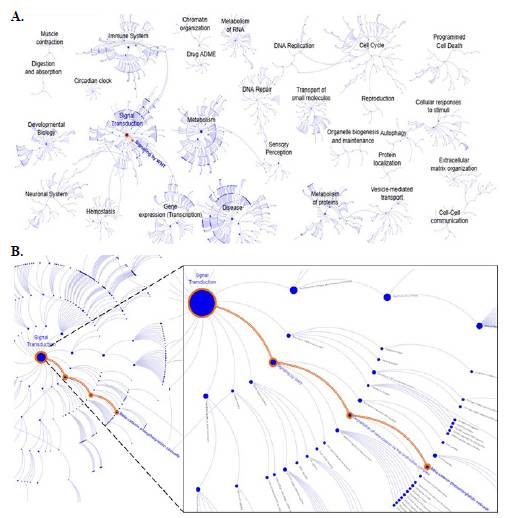
Figura 4 Caracterización de las rutas metabólicas con la herramienta Reactome utilizando el diagrama de descripción general de la ruta. A. Vías metabólicas almacenadas para Homo sapiens. B. Vía de transducción por fosforilación de la CKI, donde se observa la transducción de señales, la señalización por WNT, la degradación de la beta-catenina por el complejo de destrucción y, ya al finalizar, la cascada de fosforilación de beta-catenina.
En condiciones normales, la quinasa CKI participa en la fosforilación de la β-catenina, proteína que forma parte de un complejo de degradación, lo que conduce a su fosforilación y su posterior degradación controlada. Sin embargo, la ausencia del sitio de fosforilación en la mutación con cambio de sentido T30I afecta este proceso. Como consecuencia, la cascada de señalización Wnt/β-catenina se puede activar de una manera errónea, favoreciendo la acumulación de β-catenina y afectando la homeostasis celular.
En resumen, estos hallazgos sugieren que en la mutación T30I no solo se modifica un sitio puntual de fosforilación, sino que también se afecta de una manera significativa la regulación de la vía Wnt/β-catenina, lo que podría constituir un mecanismo molecular asociado con la aparición de cataratas en la hiperferritinemia hereditaria.
Discusión
Los niveles elevados de ferritina sérica se presentan en diversas afecciones clínicas; sin embargo, en ausencia de sobrecarga de hierro o inflamación, muchos casos quedan sin explicación (Kannengiesser et al., 2009). En este sentido, se han planteado varias hipótesis que relacionan la hiperferritinemia hereditaria con la presencia de cataratas. Entre ellas se encuentra la propuesta por Brooks et al. (2002), quienes plantean que la ferritina se encuentra en el cristalino, cuya función principal, al igual que en otros tejidos, es mantener la homeostasis del hierro. No obstante, se ha postulado que la sobreproducción de L-ferritina podría aumentar el hierro libre y, en consecuencia, las especies reactivas de oxígeno (ROS), lo que ocasiona daño oxidativo en el cristalino y, por ende, pérdida de transparencia (Brooks et al., 2002). Según Nos et al. (2021), sin embargo, está hipótesis resulta poco convincente dado que la L-ferritina no se une directamente al hierro y, además, el cristalino es pobre en este metal (Nos et al., 2021). La hipótesis más acertada hasta el momento sostiene que la acumulación de L-ferritina agregada y pobre en hierro en la corteza del cristalino origina depósitos opacos que difractan la luz y provocan cataratas bilaterales, es decir, cuando el exceso de ferritina se precipita en la corteza del cristalino forma abundantes opacidades puntiformes de color blanco, provocando la difracción de la luz e impidiendo el enfoque correcto, lo que causa cataratas bilaterales que pueden requerir la cirugía como tratamiento principal (Moravikova et al., 2020; Nos et al., 2021).
Si bien se han reportado cambios en la glicosilación de la proteína FTL asociados con la mutación T30I, incluido el sitio de glicosilación en la posición 8 (secuencia NYST) de la cadena polipeptídica, que se caracteriza por la presencia polar de la asparagina (Kannengiesser et al., 2009), en nuestro análisis bioinformático, dichos cambios no fueron significativos (Tabla 1). Este hallazgo contrasta con publicaciones previas, por lo que se sugiere que la glicosilación no constituye el eje central de la patogenia en nuestro modelo. En este sentido, la principal contribución de nuestro estudio radica en el análisis de modificaciones postraduccionales alternativas, particularmente la fosforilación.
A partir de nuestros resultados, se constató la pérdida del sitio de fosforilación S-33 en la variante T30I, el cual es reconocido por la quinasa CKI. Este hallazgo es relevante porque la CKI, en conjunto con la GSK-3 (glucógeno sintasa quinasa-3) y el complejo multiproteico integrado por el APC (gen de la poliposis adenomatosa coli supresor de tumores) y la axina, fosforilan cuatro residuos críticos de la β-catenina de serina y treonina (S-33, S-37, T-41 y S-45) de la región amino-terminal de la β-catenina, promoviendo su degradación proteosomal. La pérdida de fosforilación en S-33 puede favorecer la acumulación desregulada de β-catenina, activando de manera aberrante la vía Wnt/β-catenina, la cual desempeña un papel esencial en procesos de desarrollo a través del complejo de transcripción β-catenina-TCF/LEF, tales como la homeostasis, la formación de tejido ocular o el desarrollo de la organogénesis ocular (Amit et al., 2002; Fujimura, 2016). Además, esta desregulación se ha asociado con el cáncer colorrectal, con otras neoplasias malignas e, incluso, con las cataratas (Liu et al, 2002; Fujimura, 2016).
Se propone, entonces, que, más allá de la glicosilación, la alteración en la fosforilación mediada por la quinasa CKI constituye un mecanismo molecular alternativo y potencialmente central en la fisiopatología de la mutación con cambio de sentido T30I. Aunque nuestros resultados se basan en análisis bioinformáticos, ofrecen un marco novedoso que amplía la comprensión de los mecanismos postraduccionales involucrados y sugiere nuevas direcciones experimentales para validar la contribución de la vía de señalización Wnt/β-catenina en la aparición de cataratas en la hiperferritinemia hereditaria.
Conclusión
La hiperferritinemia con cataratas es una rara enfermedad autosómica dominante a la que se han asociado alrededor de 48 mutaciones. Entre ellas se destaca la mutación con cambio de sentido T30I en la ferritina de cadena ligera. Los estudios computacionales realizados sobre los sitios de O-glicosilación, N-glicosilación y fosforilación revelaron la ausencia de fosforilación en la serina 33 (S-33), un residuo clave en la regulación de la señalización Wnt/β-catenina, proceso fundamental durante el desarrollo ocular. La pérdida de esta fosforilación afecta la estabilidad y estructura de la FTL-wt, favoreciendo la agregación y formación de opacidades en el cristalino, dando lugar a diferentes malformaciones oculares. En particular, dicha pérdida podría explicar la aparición de las cataratas bilaterales en pacientes portadores de esta mutación. En este contexto, la fosforilación de residuos críticos como la S-33 resulta esencial para mantener la función normal de la proteína y prevenir la formación de agregados proteicos anormales. Este estudio representa una aproximación preliminar para comprender, desde una perspectiva bioquímica y bioinformática, el efecto de variantes genéticas como la T30I en la fisiopatología de la hiperferritinemia con cataratas.

