











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
En los pacientes con distrofia muscular de Duchenne (DMD) hay manifestaciones cardiovascu lares derivadas de la afectación muscular que produce el avance de la enfermedad 1. Aproximadamente un 90 % de los pacientes con DMD presentan miocardiopatía dilatada por alteraciones de la distrofina cardíaca y, en menor frecuencia, miocardiopatía hipertrófica o miocardiopatía restrictiva 1-3. Uno de los principales síntomas cardíacos en los pacientes con DMD es la falla cardíaca. Se trata de un síndrome fisiopatológico y clínico progresivo en el que el paciente presenta signos característicos asociados con trastornos circulatorios, neurohormonales y moleculares 1-3. Este síndrome se relaciona también con alteraciones cardiovasculares y extracardíacas, como edema, dificultad respiratoria, retraso ponderal e intolerancia al ejercicio 4,5.
A la fecha no existe un tratamiento curativo para la DMD ni para sus complicaciones cardiovasculares. Sin embargo, como resultado de la disponibilidad de la terapia con glucocorticoides, de la rehabilitación física, de la ventilación mecánica no invasiva y del manejo multidisciplinario con un enfoque cardiorrespiratorio y ortopédico, se ha logrado modificar la historia natural de la enfermedad 6-8 y mejorar la sobrevida de estos pacientes.
Fisiopatología de las manifestaciones cardíacas de la distrofia muscular de Duchenne
La función de la distrofina es ligar el citoesqueleto interno a la matriz extracelular; de ahí que desempeñe un importante papel en la estabilización celular 9. Al ligar el componente intracelular (actina) con el complejo glicoproteico de la membrana celular, proporciona soporte mecánico durante la contracción muscular 1,10.
La alteración en la estructura de la distrofina, secundaria a las mutaciones asociadas con la DMD, afecta la unión entre el citoesqueleto y la matriz extracelular, con posterior debilitamiento del sarcolema y la degeneración celular progresiva 1. En el corazón, la degeneración de los cardiomiocitos lleva a fibrosis 11. Inicialmente, este fenómeno aparece en la pared inferolateral y perjudica progresivamente el ventrículo izquierdo, hacia el final de la segunda década de la vida 3. Conforme aumenta la fibrosis miocárdica, hay una dilatación progresiva del ventrículo izquierdo con el respectivo incremento del trabajo cardíaco, activación del sistema renina-angiotensina y del sistema nervioso simpático 3.
Ante la alteración en la arquitectura del ventrículo izquierdo o del ventrículo derecho, cambia la remodelación ventricular (los cardiomiocitos son remplazados por fibroblastos), disminuye la función cardíaca y aparecen focos arritmogénicos 6. Además, las arritmias deterioran aún más el cuadro de falla cardíaca y se crea un círculo vicioso entre estas dos patologías. Adicionalmente, estos pacientes presentan taquicardia asociada con la disfunción del sistema nervioso autónomo. Esta falta de sincronía del ventrículo izquierdo, vinculada con una alta frecuencia cardíaca secundaria a la disfunción del sistema nervioso autónomo, contribuye a empeorar progresivamente la disfunción del ventrículo izquierdo 3.
Manifestaciones cardíacas en la distrofia muscular de Duchenne
En estadios iniciales, el compromiso cardiovascular en pacientes con DMD puede pasar desapercibido, a pesar de la disfunción cardíaca. Es más, hay un alto porcentaje de pacientes asintomáticos debido a la ausencia de actividad física por la pérdida de la deambulación 4,7,8,12. Las manifestaciones son variadas y progresivas, y se hacen notorias a los 10 años, entre las cuales la alteración de la función diastólica es el síntomas más frecuente 3,13.
Como consecuencia de los cambios en la arquitectura del corazón, el ventrículo izquierdo pierde el movimiento de torsión. La fracción de eyección se reduce en aproximadamente el 50 % que provoca una disfunción sistólica importante con hipertensión pulmonar secundaria 5,13,14. La miocardiopatía no compactada se reporta en el 28 % de los pacientes con DMD y se asocia con disminución en la función sistólica del ventrículo izquierdo, trastornos del ritmo cardíaco, falla cardíaca y eventos cerebrovasculares 13. En un menor porcentaje de pacientes, se evidencia miocardiopatía hipertrófica con disfunción diastólica temprana 15,16. A medida que avanza la cardiomiopatía, en los pacientes se producen un síndrome de bajo gasto cardíaco (signos de hipoperfusión sistémica, acidosis metabólica o mala perfusión) y un síndrome de congestión venosa sistémica o congestión venosa pulmonar, que rápidamente se convierten en una falla cardíaca global 12.
Clase funcional en falla cardíaca
La valoración de la clase funcional es compleja en pacientes con DMD, debido a que los factores evaluados no se encuentran validados para estos pacientes 17. Esto hace que las clasificaciones disponibles no sean las más adecuadas para hacer el seguimiento clínico de la clase funcional en los pacientes con DMD. En adolescentes y adultos es posible usar la clasificación de la New York Heart Association (NYHA) o la escala de la American College of Cardiology/American Heart Association Task Force (ACC/AHA) 18-20, sin que sean específicas para pacientes con DMD. En niños también es posible el uso de la escala de clasificación de Ross, que determina una valoración global de la severidad de la falla cardíaca, aunque esta escala tampoco es específica para pacientes con DMD 21,22 (tabla 1). En general, se puede utilizar la clasificación por estadios ACC-AHA en estos pacientes (tabla 2) 23.
Tabla 1 Clasificación de NYHA y clasificación de Ross
| Clase | New York Heart Association 19 | Ross 22,24 |
|---|---|---|
| I | • No limitaciones en la actividad física | • Asintomático |
| II | • Ligera limitación de la actividad física • Asintomático en reposo, la actividad física habitual causa disnea, cansancio, palpitaciones o angina | • Taquipnea leve o sudoración con la alimentación en lactantes • Disnea con el ejercicio en niños mayores |
| III | • Limitación marcada de la actividad física • Asintomático en reposo, pero actividades menores causan síntomas | • Marcada taquipnea o sudoración con la alimentación en lactantes • Tiempo de tomas prolongado con escasa ganancia ponderal • Marcada disnea con el ejercicio en niños mayores |
| IV | • Incapacidad de cualquier actividad física sin síntomas • Los síntomas están presentes incluso en reposo y con cualquier actividad se incrementan | • Síntomas en reposo: taquipnea, sudoración, retracciones |
Tabla 2 Clasificación por estadios según el American College of Cardiology y la American Heart Association 20
| Estadio A Riesgo de desarrollar insuficiencia cardíaca con función cardíaca normal y sin cambios en las cavidades por la sobrecarga de volumen | Estadio B Morfología cardíaca anormal y sin síntomas de insuficiencia cardíaca |
| Estadio C Cardiopatía estructural o funcional de base con síntomas de insuficiencia cardíaca | Estadio D Insuficiencia cardíaca en estadio terminal que requiere ionotrópicos, soporte mecánico circulatorio y trasplante cardíaco |
Fuente: tomado y adaptado de Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44(1):8-19.
Evaluación de las manifestaciones cardíacas y seguimiento en la distrofia muscular de Duchenne
Dada la variabilidad del espectro clínico y las difusas manifestaciones en pacientes con DMD, es necesario un diagnóstico temprano que establezca una terapia precoz y adecuada, así como intervenciones antes de que aparezca la insuficiencia cardíaca 4,7,8,12,25. Esto es fundamental para mejorar la calidad de vida y maximizar la supervivencia de los afectados. El objetivo es entonces abordar tempranamente a los pacientes con DMD, en el estadio preclínico, para poder determinar por ecocardiografía, Strain, resonancia, electrocardiograma y biomarcadores, la presencia de disfunción diastólica 12,24-27.
En principio, se recomienda una evaluación cardiovascular integral, en el momento del diagnóstico de DMD, que incluya valoración clínica por cardiología pediátrica, electrocardiograma basal, imágenes no invasivas como ecocardiografía (examen de elección en estos pacientes) y resonancia magnética nuclear cardíaca, que permiten evaluar la viabilidad miocárdica, determinar zonas de fibrosis y evaluar puntajes de riesgo 12,24,25,27. Sin embargo, pueden presentarse limitaciones para realizar estos exámenes relacionadas con su costo y disponibilidad 20,28. Nuevas técnicas ecocardiográficas como el Strain permiten detectar tempranamente cambios mínimos en la función sistólica y diastólica, incluso por debajo de las edades establecidas 29,30.
El seguimiento del paciente con DMD debe ser integral, óptimo y multidisciplinario, con recomendaciones que varían de acuerdo con la fase en la que se encuentra (tabla 3). Se requiere la toma de exámenes paraclínicos y estudios imagenológicos, entre los cuales se encuentran 31-34:
Laboratorios: hemograma, proteína C reactiva, enzimas cardíacas (troponina), electrolitos, perfil de infección, cistatina C (un marcador de función renal no influenciado por la degradación del músculo esquelético) y pruebas de función hepática (que pueden estar alteradas por congestión venosa sistémica).
Biomarcadores: para el seguimiento de los pacientes, se recomienda la toma de péptido natriurético tipo B (su uso debe ser racional e individualizado). Valores >300 pg/ml son un predictor de hospitalización por insuficiencia cardíaca o mal pronóstico 20.
Radiografía de tórax: para definir alteraciones propias de la enfermedad de base o comorbilidades.
Electrocardiograma: puede evidenciar un ritmo sinusal con algunos trastornos en la repolarización o aumento en la amplitud de la onda R, extrasístoles ventriculares o supraventriculares y salvas de taquicardia ventricular 35.
Ecocardiografía: debe incluir fracción de eyección, fracción de acortamiento, desplazamiento sistólico del plano del anillo tricúspideo, examen Doppler pulsátil, continuo y tisular, y el uso de Strain, para medir la deformación en los distintos segmentos cardíacos y evaluar mejor la función cardíaca.
Tabla 3 Seguimiento y diagnóstico de pacientes con distrofia muscular de Duchenne 34
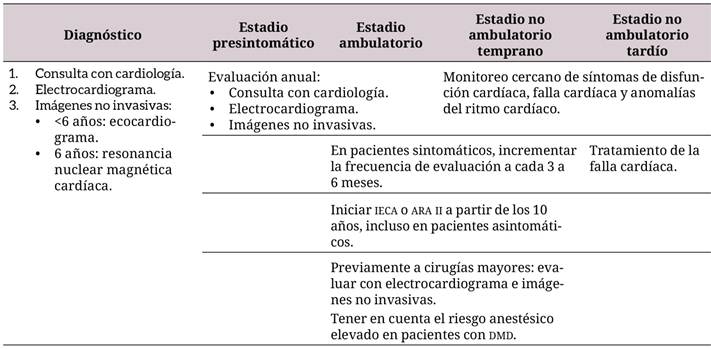
IECA: enzima convertidora de la angiotensina; ARA II: antagonista de los receptores de la angiotensina n.
En pacientes con DMD asintomáticos desde el punto de vista cardiovascular, se recomienda la toma de un ecocardiograma al diagnóstico y hacia los 6 años de vida, con un seguimiento anual hasta los 10 años. Luego, según la afectación cardiovascular, los controles deben ser más frecuentes (cada 3 a 6 meses), con evaluaciones clínicas y paraclínicas (incluyendo electrocardiograma, ecocardiograma, Holter y resonancia magnética nuclear cardíaca) que permitan identificar el momento ideal para la intervención y el establecimiento del tratamiento 25,28,34,36,37.
Tratamiento de las manifestaciones cardíacas en la distrofia muscular de Duchenne 23,27,31
Las terapias establecidas para tratar los efectos musculares de la enfermedad en pacientes con DMD son inefectivas para tratar el compromiso cardiovascular 27,28,34. El manejo farmacológico de la insuficiencia cardíaca se basa en diuréticos, inhibidores de la enzima convertidora de la angiotensina y betabloqueadores, de acuerdo con el estadio del paciente (tabla 4) 5,28,38.
Tabla 4 Tratamiento de la insuficiencia cardíaca en pacientes con DMD, de acuerdo con la estratificación 3,26,39
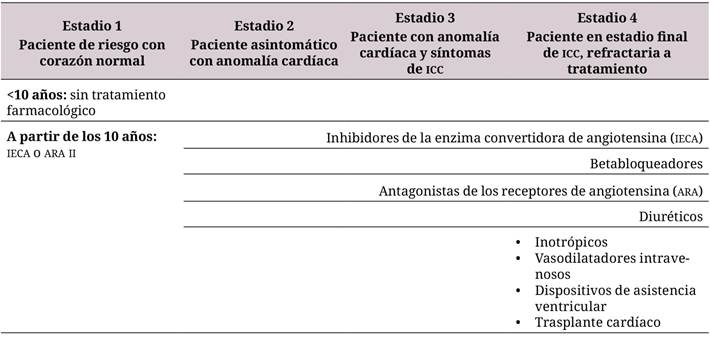
ICC: insuficiencia cardíaca congestiva; IECA: inhibidores de la enzima convertidora de angiotensina; ARA: antagonistas de los receptores de angiotensina.
Inhibidores de la enzima convertidora de la angiotensina
Son unos de los medicamentos más utilizados y la primera línea de manejo en pacientes con DMD 1. Los inhibidores de la enzima convertidora de angiotensina (IECA) se usan para enlentecer la remodelación cardíaca que precede al daño del cardiomiocito. Disminuyen la resistencia vascular periférica, mejoran la función endotelial y retrasan la poscarga 1,38.
Las guías actuales recomiendan iniciarlos en pacientes asintomáticos a los 10 años o antes, según la evolución 38,40. Lo anterior se basa en la capacidad de los IECA de retrasar la progresión de la enfermedad cardíaca en la DMD con fracción de eyección normal, mejorando así la sobrevida y calidad de vida de los pacientes 12,23,41.
Inhibidores del receptor de la angiotensina
Los inhibidores del receptor de la angiotensina son la alternativa al uso de IECA, según el tipo de paciente y su edad 1,5,38. Tienen la habilidad de prevenir el daño del cardiomiocito en momentos de esfuerzo cardíaco 38.
Betabloqueadores
Los betabloqueadores se utilizan en combinación con los IECA y usualmente se indican en pacientes con taquicardia o arritmias 38,39, dada la disfunción autonómica en el corazón distrófico de los pacientes con DMD 35. Los estudios de los betabloqueadores como monoterapia en DMD son controversiales, con poca evidencia de lograr efectos positivos importantes 38. Sin embargo, su uso es avalado por la Sociedad Europea de Cardiología, como manejo concomitante con IECA, en pacientes con disfunción sistólica, pues reducen la mortalidad y la tasa de hospitalización 18.
Otras moléculas
La Sociedad Española de Cardiología recomienda el inicio de diuréticos en caso de sobrecarga de líquidos en pacientes sintomáticos o en pacientes refractarios al tratamiento 40. Dentro de los diuréticos, el más usado es la furosemida, como primera línea, y los diuréticos tiazídicos, como medicamentos de segunda línea 40.
En el manejo de la insuficiencia cardíaca en pacientes con DMD, las dosis de los tales medicamentos son similares a las administradas a otros pacientes con insuficiencia cardíaca y miocardiopatía no secundaria a DMD (tabla 5) 3,26,39.
Tabla 5 Dosis de medicamentos para el manejo de la insuficiencia cardíaca en pacientes con distrofia muscular de Duchenne 3,26,39
| IECA | Betabloqueadores | Diuréticos/antagonistas de la aldosterona |
| Enalapril: • Dosis pediátrica (máxima): 0.1-0.4 mg/kg cada día • Niños < 50 kg: 10 mg/día • Niños > 50 kg: 20 mg/día | Carvedilol: • Dosis pediátrica (máxima): 0.2-1 mg/kg cada día • Niños < 50 kg: 25 mg/día • Niños > 50 kg: 50 mg/día | Furosemida: • Dosis pediátrica (máxima): 1-2 mg/kg dosis, cada 6 a 24 horas • Niños > 50 kg: 20-80 mg/día |
| Captopril: • Dosis pediátrica (máxima): 0.5 3 mg/kg cada día • Niños > 50 kg: máximo 100 mg/ día | Metoprolol: • Dosis pediátrica (máxima): 1-2 mg/kg cada día • Máximo 100 mg/día en ambos rangos de peso | Espironolactona: • Dosis pediátrica (máxima): 1-3.3 mg/kg cada día • Incrementar hasta 5-6 mg/kg cada día • Dosis dividida en cada 6 a 24 h • Dosis máxima: 100 mg/día |
IECA: inhibidores de la enzima convertidora de angiotensina.
Algunos estudios sugieren que los antagonistas de receptores de mineralocorticoides, por ejemplo, la eplerenona, son eficaces para atenuar la disminución de la función sistólica del ventrículo izquierdo 42,43. Cabe resaltar que la disponibilidad y uso de tratamientos específicos para los problemas cardíacos es de vital importancia en estos pacientes.
Conclusiones
El diagnóstico y el tratamiento precoz de la DMD y la cardiomiopatía, al igual que el seguimiento estrecho y el manejo integral, óptimo y multidisciplinario, son fundamentales para mejorar la calidad de vida y maximizar la supervivencia de los pacientes.
Es ideal realizar una evaluación cardiovascular integral en el momento del diagnóstico de DMD que incluya la valoración clínica por cardiología pediátrica o de adultos, electrocardiograma, ecocardiografía y resonancia magnética nuclear cardíaca. Posteriormente, se debe hacer un seguimiento mediante evaluación cardíaca integral que incluya electrocardiograma, ecocardiograma y biomarcadores. Esta evaluación debe ser anual para los pacientes asintomáticos y cada 3 a 6 meses cuando aparecen alteraciones morfológicas o funcionales.
Se recomienda administrar un IECA o un antagonista de los receptores de la angiotensina en los niños con DMD, comenzando a los 10 años, o antes, según su evolución, para retrasar la progresión de la enfermedad cardíaca.
La insuficiencia cardíaca manifiesta en pacientes con DMD y se trata de acuerdo con las guías de manejo de falla cardíaca.

