











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
Los paragangliomas son tumores neuroendocrinos derivados de las porciones simpáticas y parasimpáticas de los paraganglios del sistema nervioso autónomo. Su prevalencia anual ajustada por edad en Estados Unidos es alrededor de 0,3 a 0,7 por cada 1.000.000 de habitantes (1). La edad de inicio de los casos esporádicos es de aproximadamente 40 a 50 años y para casos hereditarios es generalmente antes de los 40 años (2).
Cuando los paragangliomas se encuentran en la región toraco-abdomino-pélvica, son secretores de hormonas en el 85 % de los casos; mientras que no son secretores en un 90 % de los casos, cuando se localizan en la cabeza y cuello (3). Cuando se presentan en la cabeza y el cuello, la localización más frecuente es el cuerpo carotídeo. Los paragangliomas del cuerpo carotídeo usualmente tienen un comportamiento benigno, y solo en el 6 %-12 % de los casos se documenta metástasis (4).
En general, la malignidad no se determina por estudios histológicos; se requiere la presencia de un compromiso metastásico, habitualmente, a los ganglios linfáticos, los huesos, el hígado y los pulmones (5). Las alteraciones somáticas en los genes implicados en la preservación de los telómeros e inmortalización celular (es decir, ATRX o TERT) están relacionadas con la presencia metastásica (6, 7). Dado lo anterior, la activación de la telomerasa y las mutaciones de ATRX podrían usarse para identificar paragangliomas metastásicos, sobre todo en tumores con alto riesgo de progresión, como aquellos con un tamaño mayor a 5 cm o con mutación en la succinato deshidrogenasa tipo B (8).
En los últimos años, se han identificado más de 15 genes de susceptibilidad; la mayoría son genes supresores de tumores (incluidos NF1, VHL, SDHx, FH, TMEM127, MAX o SCL25A11) y algunos son oncogenes (RET y EPAS1) (9). Una mutación de la línea germinal en uno de estos se encuentra en el 40 % de todos los casos, lo que hace que el paraganglioma sea el tumor más heredado en humanos (10). La mayor parte de los paragangliomas de cuerpo carotídeo presentan mutaciones de la SDHx, lo cual está asociado con la presencia de enfermedad más agresiva y menos supervivencia libre de enfermedad luego de la intervención (11).
Caso clínico
El caso corresponde a un hombre de 34 años sin antecedentes personales o familiares relevantes, quien en la consulta presentaba un cuadro de dolor torácico pleurítico, tos seca, dolor en la región inguinal derecha, limitación para la movilización del miembro inferior ipsilateral y pérdida involuntaria de 15 kg de peso en el último mes. En el examen físico presentaba signos vitales normales y dolor con los arcos de movilidad de la cadera derecha.
En la tomografía de tórax se documentó una masa en el tercio inferior del pulmón izquierdo que originó la erosión de las vértebras adyacentes e infiltración de la pleura parietal. Así mismo, las radiografías de pelvis y cadera derecha mostraron una fractura impactada subcapital del fémur derecho y lesiones líticas en todas las estructuras óseas visualizadas.
Como estudio de extensión, la tomografía de abdomen mostró un hígado aumentado de tamaño, de contornos nodulares, masas redondeadas con necrosis, además de un conglomerado ganglionar paraaórtico izquierdo de 52 × 49 mm y glándulas suprarrenales de aspecto normal. La biopsia percutánea de pulmón mostró un tumor epitelioide con ligera atipia (figura 1A) con cambio focal de célula clara. Entre tanto, los estudios de inmunohistoquímica evidenciaron negatividad para citoqueratinas con expresión de marcadores neuroendocrinos, como son la cromogranina y sinaptofisina (figura 1B). Hubo reactividad para S100 en patrón sustentacular con Ki67 del 5 %. Los hallazgos de un tumor neuroendocrino con negatividad para marcadores epiteliales y patrón periférico del S100 corresponden a un paraganglioma. Las concentraciones de metanefrinas libres en el plasma fueron normales: 72,1 pg/ml (valor de referencia: <90 pg/ml) y los de cromogranina A fueron mayores de 700 µg/L (valor de referencia: <100 µg/L).
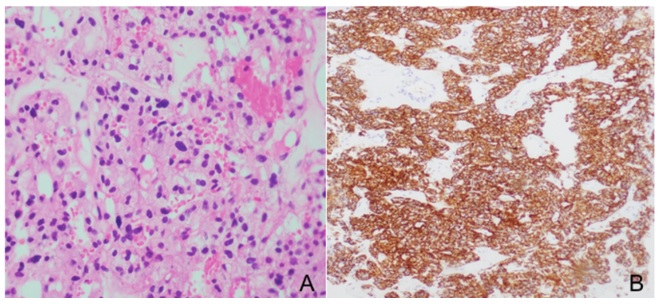
Figura 1 A) Pulmón con tumor epitelioide. A 40X se observan nidos con ligera atipia y cambio de célula clara. B) Sinaptofisina positiva de forma difusa
Por presencia de dolor lumbar con signos de alarma, se llevó a resonancia de columna, que mostró un extenso compromiso metastásico de toda la columna sin mielopatía compresiva. Se encontró una masa en la bifurcación carotídea izquierda que separaba el origen de las carótidas internas y externas, de 26 × 22 × 20 mm, compatible con paraganglioma, concordante con los hallazgos de la patología.
Durante su estancia en sala general, presentó elevación de las transaminasas (5 veces el límite superior) asociado con episodios recurrentes de hipoglucemia sintomática con concentraciones de cortisol 8 am dentro de límites normales (20,56 µg/dL) y requerimiento persistente de dextrosa y dexametasona, que limitó ofrecer algún tratamiento específico.
El paciente fue valorado por el servicio de genética clínica, que indicó un panel molecular para MAX, NF1, RET, SDHA, SDHAF2, SDHB, TMEM127 y VHL. Sin embargo, tales estudios no se realizaron, por la muerte precoz del paciente a los 15 días del diagnóstico inicial, por falla hepática.
Discusión
Los paragangliomas presentan alta variabilidad en su presentación clínica, por su localización, potencial de metástasis, fenotipo bioquímico y predisposición genética. Resaltamos este caso poco frecuente de paranganglioma carotídeo, en quien se pensó inicialmente tendría un tumor de origen pulmonar, con hallazgos de compromiso metastásico extenso. En este hombre, al igual que lo reportado en la literatura, no se observaron síntomas de producción de catecolaminas, dado que los paragangliomas de cabeza y cuello, rara vez, las producen de forma excesiva (5 %) y su presentación clínica está dada, sobre todo, por síntomas relacionados con el efecto de masa por compresión de tejidos adyacentes y nervios craneales.
Actualmente, cerca del 40 % de los casos presentan mutaciones relacionadas con síndromes hereditarios. Algunos datos aumentan la probabilidad pretest de un síndrome hereditario, por ejemplo: hallazgos clínicos sugestivos de neurofibromatosis, edad menor de 35 años, tumores sincrónicos y malignidad (12). En nuestro caso, la edad de 34 años y la presencia de malignidad fueron nuestros datos cardinales para solicitar la valoración por parte del servicio de genética clínica, a fin de descartar los síndromes hereditarios relacionados con paragangliomas, como la neoplasia endocrina múltiple tipo 2, neurofibromatosis tipo 1, enfermedad de Von Hippel Lindau y los paragangliomas hereditarios.
La mayoría de paragangliomas se identifican por medio de tomografía axial computarizada y resonancia magnética nuclear, pues ambos estudios tienen una sensibilidad superior al 90 % y una especificad superior al 75 %. Dentro de los estudios funcionales se encuentra la gammagrafía con meta-yodo-bencil-guanidina (MIBG) y la tomografía por emisión de positrones con 18F-3,4 dihidroxifenilalanina. Sin embargo, su rol actual no está claramente establecido, dado su alto costo, poca disponibilidad y que solo detectan un 1,5 % más de tumores primarios y un 3,5 % más de tumores metastásicos. Se reserva su toma para pacientes con predictores de compromiso metastásico, como: tamaño tumoral mayor a 5 cm y la presencia de una mutación en la succinato deshidrogenasa tipo B (8,13).
Aunque la mayoría de los paragangliomas son curados con cirugía, en algunas ocasiones (15 %-20 %) hay evidencia de enfermedad avanzada por afectación metastásica (14); en el caso de este paciente, en quien se documentó un paraganglioma del cuerpo carotídeo con afectación metastásica múltiple se resalta lo infrecuente de dicha presentación. La terapia sistémica es una opción de tratamiento que incluye quimioterapia citotóxica y la terapia con 131-I MIBG para pacientes con trasportadores de recaptación de noradrenalina (15). La evolución clínica tórpida y el extenso compromiso hepático impidieron el inicio de la quimioterapia, sumado a que no se disponía en su momento de terapia con 131-I MIBG en la institución.
Hoy en día, las opciones de tratamiento para paraganglioma metastásico son escasas, y estos pacientes tienen una sobrevida corta del 60 % a 5 años. Clásicamente, se ha usado la quimioterapia con esquema CVD (ciclofosfamida, vincristina, dacarbazina) para el manejo de la enfermedad metastásica; sin embargo, solo cerca del 40 % logra tener respuesta parcial a esta terapia (16).
En el momento hay estudios en curso para evaluar el uso de nuevas terapias para el paraganglioma metastásico, como el uso de terapia con inhibidores de tirosina-cinasa (por ejemplo, sunitinib, pazopanib, cabozantinib y axitinib), al igual que el uso de inhibidores de la ruta mTOR (por ejemplo, el everolimus) e inmunoterapia (por ejemplo, pembrolizumab), que en el futuro próximo se espera sean alternativas de tratamiento para los pacientes con paraganglioma metastásico (17,18).
Los paragangliomas de cabeza y cuello son una entidad rara con comportamiento usualmente benigno. El diagnóstico es tardío, por la falta de síntomas relacionados con la producción de catecolaminas y a que se requiere la presencia de metástasis para confirmar su malignidad. Es necesaria la búsqueda activa de producción de catecolaminas y asesoramiento genético, por la alta frecuencia de mutaciones germinales.

