











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Abreviaturas
AIC: aneurisma intracerebrales.
AMPc: adenosín monofosfato cíclico
CA 19-9: antígeno de cáncer 19-9
EPO: eritropoyetina
ERC: enfermedad renal crónica
ERC E5: enfermedad renal crónica estadio 5
HSA: hemorragia subaracnoidea
HTA: hipertensión arterial
H VI: hipertrofia ventricular izquierda
ITU: infección del tracto urinario
LDL: lipoproteínas de baja densidad
NHS: National Health Service
ON: óxido nítrico
PQRAD: poliquistosis renal autosómica dominante
RMN: resonancia magnética nuclear
SNS: sistema nervioso simpático
SRAA: sistema renina-angiotensina-aldosterona
TC: tomografía computarizada
TFGe: tasa de filtración glomerular estimada
VI: ventrículo izquierdo
VRT: volumen renal total
Introducción
La poliquistosis renal autosómica dominante (PQRAD) es la causa más frecuente de nefropatía hereditaria en el adulto y de enfermedad renal crónica estadio 5 (ERC E5) de origen genético [1, 2]. Se caracteriza por el desarrollo de quistes renales que se presentan desde etapas tempranas de la vida, aunque las complicaciones clínicas tienden a manifestarse en etapas más tardías en la tercera a cuarta década de la vida. Estos quistes se desarrollan de manera progresiva hasta afectar completamente la arquitectura del parénquima renal (cistogénesis) y lleva a la pérdida de la función renal entre la quinta y la sexta década de la vida. Se estima que el 45-70 % de los pacientes con PQRAD progresan a ERC E5 a la edad de 65 años [2].
La PQRAD es una enfermedad muy variable en términos de resultados clínicos, progresión renal, complicaciones y tratamiento, y aunque principalmente su compromiso es renal, la PQRAD es una enfermedad multisistémica con implicaciones más allá de la salud renal del individuo [2, 3]. Sus manifestaciones sistêmicas comprenden: hipertensión arterial, aneurismas intracraneales, poliquistosis hepática, anomalías valvulares cardiacas y quistes en otros órganos [4].
Este trastorno genético ha afectado a alrededor de 12,5 millones de personas en todo el mundo, independientemente del sexo y del origen étnico. Además, es responsable de alrededor del 10 % de los pacientes que desarrollan ERC E5, los cuales pueden requerir diálisis o trasplante renal [1,5,6].
La tasa de prevalencia de PQRAD es variable entre los diferentes artículos revisados, encontrando reportes de tasas de 1 en 400 habitantes a 1 en 2000 habitantes, esta disparidad se debe posiblemente a fallas de registro, progresión y gravedad, lo cual varía notablemente entre los pacientes [5-7].
Se ha reportado para Colombia una prevalencia de PQRAD de 9,81 por cada 100 000 habitantes, con 4,35 casos de terapia de sustitución renal presentada en una media de 52,58 años. Estos datos suministrados por la Cuenta de Alto Costo [8] son muy inferiores a lo reportado por otros países como Brasil con una prevalencia de 1:190 habitantes [9], Estados Unidos, con 182,1 a 212,4 casos por cada 100 000 habitantes, con incidencia de 5000 casos al año, Francia con 1 en 1111, China con 1 en 1130 y Japón con 1 en 4033 [10]. Dada la gran diferencia de reporte de casos, cabe suponer que existe un alto subregistro de este diagnóstico en Colombia, dado que, al tener en cuenta el promedio de prevalencia mundial estimado en 1 por cada 2000 personas, con la actual población colombiana se estimaría que debería haber aproximadamente 25 000 personas con PQRAD.
Los dos principales genes alterados en la PQRAD son PKD1, localizado en el cromosoma 16 en su brazo corto 16p13.3 y PKD2, localizado en el cromosoma 4 en su brazo largo 4q21, dando como resultado alteraciones en las proteínas policistina-1 (PC1) y policistina-2 (PC2), respectivamente, donde la primera es más común y representa el 78 % de los casos, cuya presentación es una enfermedad con clínica severa y de peor pronóstico, mientras que la segunda representa el 15 % de los casos [1,4,5].
Se han identificado otros genes causantes de la enfermedad (GANAB, DNAJB11), en familias con poliquistosis renal leve y poliquistosis hepática variable, donde las mutaciones en el gen GANAB representan aproximadamente el 0,3 % del total de casos de PQRAD. Además, el gen DNAJB11 se caracteriza por la presencia de poliquistosis renal, enfermedad renal tubulointersticial autosómica dominante con riñones quísticos de tamaño normal y fibrosis intersticial progresiva que representa el 0,1 % de los casos [4, 6, 11, 12].
Otros genes asociados menos comunes (ALG5, ALG9, IFT140), identificados mediante pruebas de genética molecular, explican un pequeño número de casos con sospecha clínica de PQRAD [13].
A la fecha de sometimiento de este documento se han informado 2322 mutaciones del gen PKD1 y 278 del gen PKD2 en la base de datos de mutaciones de la enfermedad renal poliquística a través de la herramienta desarrollada y mantenida por la Mayo Clinic (https:Zpkdb.mayo.edu/variants), que incluye todas las variantes de secuencia descritas en estos genes [5,14].
Los hijos de padres afectados tienen un 50 % de probabilidad de adquirir la mutación del gen y cerca del 5 % al 10 % de los casos aparece como una mutación espontánea [15].
La enfermedad presenta diferentes velocidades de evolución, donde la variabilidad genética influye en la gravedad de la enfermedad, encontrando que las mutaciones truncadas de PKD1 están asociadas con una edad más temprana de aparición de la enfermedad renal (edad media de 58 años) que las mutaciones no truncadas de PKD1 (edad media de 67 años) y las mutaciones PKD2 (edad media de 79 años) [16].
Fisiopatología
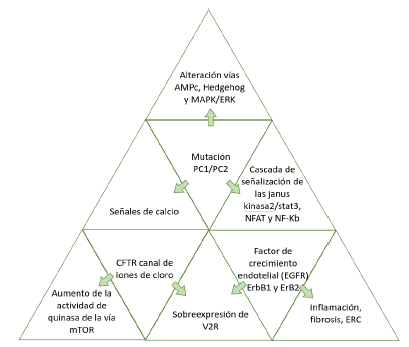
La patología de la PQRAD es compleja e implica el mal funcionamiento de diferentes vías de señalización como AMPc, Hedgehog y MAPK/ERK, debido a la mutación de policistina-1 o policistina-2.
A nivel renal, la policistina-1 se encuentra particularmente en la nefrona distal, en los cilios primarios de las células epiteliales del conducto colector, donde su función es compleja y se ha propuesto como activador de la cascada de señalización de la janus kinasa2/STAT3, NFAT y NF-Kb [15]; mientras que la policistina-2 es un canal de iones permeable al calcio que se encuentra en la membrana del retículo endoplásmico (RE) y regula la liberación de iones de calcio desde el RE, proliferación y diferenciación celular. Además, la policistina-2 también se encuentra en la membrana celular basolateral de células epiteliales tubulares [1], donde ambas proteínas se localizan en los cilios primarios, presentes en casi todas las células del organismo.
La cistogénesis en la poliquistosis renal se genera cuando tanto las copias de PC1 o PC2 mutada causan el defecto en los cilios primarios (ciliopatía). La expresión abrupta resultante (subexpresión o sobreexpresión) de policistina-1 o policistina-2 conduce a la interrupción de varias vías de señalización intracelular, las cuales conllevan al desarrollo progresivo de quistes, debido a la desregulación de la proliferación celular y la secreción de líquidos en los quistes [1]. La pérdida de PC1 o PC2 conduce a una disminución en el calcio intracelular y posteriormente a una respuesta celular anormal de la adenilciclasa, aumentando los niveles de adenosín monofosfato cíclico (AMPc), esto promueve el aumento de la secreción de líquidos y la proliferación de células epiteliales.
Se ha propuesto que la cistogénesis en PQRAD incluye dos fases: iniciación del quiste y expansión del quiste, mediado por la pérdida o reducción de la función de PC1 o PC2 [17], con la expansión de los quistes se lesiona el tejido circundante, causando inflamación y fibrosis con progresión del daño renal [1]
Las moléculas de señalización incluidas AMPc, mTOR, AMPK y los factores de crecimiento, se han implicado en la patogénesis de poliquistosis renal, donde la PC1 regula indirectamente el aumento de la actividad quinasa de la vía mTOR (por sus siglas en inglés: mamalian target ofrapamycin) que promueve la hipertrofia, división celular y supervivencia celular [10,18].
El AMPc es uno de los componentes clave en el crecimiento quístico renal, ya que activa la proteína quinasa extracelular por vía de la quinasa regulada por señales (ERK). El receptor selectivo de vasopresina tipo 2 (V2R) se expresa principalmente en las células del conducto colector de la nefrona, donde la sobreexpresión de V2R incrementa las concentraciones de AMPc y de los receptores del factor de crecimiento endotelial (EGFR) ErbB1 y ErbB2 en la región apical, factor que estimula la cadena de señalización MAPK/ERK [15]. En este proceso, el papel de la vasopresina en la PQRAD es sustancial.
Otra vía de señalización implicada es la Hedgehog (Hh), la cual regula la proliferación celular, diferenciación y polaridad tisular en los cilios primarios.
El canal de iones de cloro (CFTR) está codificado por el gen CFTR, cuya mutación se ha relacionado con la secreción de líquido por el epitelio de paredes del quiste [15]. La figura 1 resume los mecanismos implicados en la citogénesis.
Manifestaciones clínicas renales y extrarrenales de la PQRAD
La PQRAD es una enfermedad multisistémica, cuya sintomatología es de inicio tardío, caracterizado por la formación de quistes renales que se forman desde etapas tempranas de la vida, aunque las complicaciones se manifiestan en la tercera y cuarta década de la vida [13]. El aumento del tamaño renal es progresivo y conlleva a enfermedad renal, donde aproximadamente el 50 % de los pacientes con PQRAD desarrollan ERC E5 [19]. La PQRAD está asociada a anormalidades que afectan otros órganos: como quistes en el hígado, vesículas seminales (40 %), páncreas, membrana aracnoidea (8 %), bazo, desarrollo de divertículos de colón y anomalías vasculares (aneurismas intracraneales, dilatación de la raíz aortica y disección de la aorta torácica) [6,12,13].
La hipertensión arterial, la disfunción endotelial, la inflamación sistémica y la aterosclerosis acelerada son alteraciones que se encuentran en etapas tempranas de la enfermedad y son responsables de aumentar, tanto los riesgos cardiovasculares, como la progresión hacia ERC E5 [20]. En la tabla 1 se resumen las manifestaciones clínicas de la PQRAD, su frecuencia de aparición, los mecanismos implicados y sus consecuencias.
Tabla 1 Manifestaciones clínicas
| Manifestación clínica | Frecuencia de aparición | Mecanismo | Consecuencias |
|---|---|---|---|
| Hipertensión arterial | 50-75 % [20] |
|
|
| Proteinuria /albuminuria | Sin dato | • HTA no controlada y aumento del riesgo de proteinuria [13,22] | • Marcador de gravedad de la enfermedad [13] |
| Quistes hepáticos |
|
|
|
|
Cardiacas:
|
|
|
|
| Litiasis |
|
|
|
|
Anormalidades vasculares:
|
|
La edad media de rotura de los aneurismas intracraneales es menor en los individuos con PQRAD que en la población general (39 años frente a 51 años) |
|
|
Alteraciones hematológicas
|
|
|
• Linfopenia, mediador de un mayor riesgo de infección (tracto urinario y respiratorio) y malignidad [24,32,33] |
| Pulmonares: Bronquiectasias |
|
|
• Mayor riesgo de infección |
| Alteraciones en el metabolismo de la glucosa | • Riesgo de desarrollar 2 a 3 veces diabetes después del trasplante [33,35,36] |
|
• Riesgo de desarrollar diabetes |
| En investigación bajo recambio óseo | Sin datos | Fenotipo óseo particular que es atribuible al papel de la policistina en la fisiología del osteocito [24,37] | • Precaución con el uso de bifosfonatos [24] |
Nota: AIC: aneurisma intracerebrales, EPO: eritropoyetina, HSA: hemorragia subaracnoidea, HTA: hipertensión arterial, HVI: hipertrofia del ventrículo izquierdo, ON: óxido nítrico, PQRAD: poliquistosis renal autosómica dominante, SNS: sistema nervioso simpático, SRAA: sistema renina angiotensina aldosterona, VI: ventrículo izquierdo, VRT: volumen renal total.
Fuente: elaboración propia.
Complicaciones que se pueden presentar en el paciente con PQRAD: renales y extrarrenales
La tabla 2 describe las complicaciones de la PQRAD.
Tabla 2 Complicaciones de la PQRAD
| Complicación | Frecuencia de aparición | Datos relevantes |
|---|---|---|
| Dolor | Relacionado con el tamaño de los quistes [6] | Dolor lumbar y abdominal Obesidad: mayor riesgo de dolor lumbar y radicular [38] Las etiologías del dolor: hemorragia del quiste, nefrolitiasis, infección del quiste y (raramente) tumor. |
| Infección urinaria | Presente en 30-70 % [24] de los pacientes | Pielonefritis: más común en pacientes con ERC E5 por PQRAD (16 %) [24] Los factores anatómicos están implicados en la predisposición a ITU |
| Hemorragia quística | Presente hasta en un 60 % [6] | Hematuria Dolor Infección Descenso de valor de hemoglobina |
| Infección quística | Del 30 % al 50 % de los pacientes con PQRAD pueden desarrollar al menos una infección en algún momento durante su vida. El 10 % ameritará hospitalización [39] | Dolor Fiebre PCR >5 mg/dl La elevación de fosfatasa alcalina y CA 19-9 [4] |
| Evento coronario | Los pacientes con PQRAD tienen mayor frecuencia de IAM [25] El IAM en pacientes con PQRAD en América se presenta hasta en un 6 % [25] | Muerte |
| Ruptura de aneurisma cerebral |
|
Consecuencias:
|
| Complicaciones cardiovasculares | Causa más común de muerte en PQRAD [25] | Inicio temprano de HTA, edad promedio: 27 años [21] |
Nota: AIC: aneurisma intracebral; CA 19-9: antígeno de cáncer 19-9; ERC E5: enfermedad renal crónica estadio 5; HSA: hemorragia subaracnoidea; HTA: hipertensión arterial; ITU: infección del tracto urinario; PQRAD: poliquistosis renal autosómica dominante; RMN: resonancia magnética nuclear; PCR: Proteína C reactiva; IAM: Infarto agudo de miocardio.
Fuente: elaboración propia.
Materiales y métodos
Objetivo
Generar recomendaciones basadas en la evidencia para el diagnóstico, seguimiento y tratamiento de la PQRAD, a través de un consenso de expertos.
Población objetivo del consenso
Pacientes con sospecha o diagnóstico de PQRAD, pacientes que se encuentran en proceso de trasplante por esta enfermedad o que se encuentran interesados en asesoramiento genético de la misma para tratamientos de fertilización o planificación familiar, así como familiares de pacientes con esta enfermedad e interesados en ser donantes de órganos para trasplante.
Aspectos clínicos
Se brinda una contextualización clínica referente a la definición, la epidemiología, las características clínicas, el diagnóstico diferencial y las complicaciones de la enfermedad. El consenso se centró en preguntas referentes a diagnóstico, tratamiento, seguimiento, identificación de progresión de la enfermedad y recomendaciones referentes al trasplante renal y la asesoría genética.
Metodología
Se generaron las preguntas de interés por parte del grupo coordinador, conformado por una especialista en Nefrología, quien fue la líder desarrolladora del consenso y un médico epidemiólogo experto en la metodología, estos profesionales fueron seleccionados por su experiencia en el tema y brindaron el soporte técnico y metodológico del consenso a desarrollar.
Grupo desarrollador
La Asociación Colombiana de Nefrología e Hipertensión Arterial (Asocolnef) convocó a un total de 10 especialistas en Nefrología, de acuerdo con su experiencia para realizar el presente consenso, con la participación de diferentes zonas del país como el eje cafetero, Caribe, Oriental, Centro y Pacífico, con el fin de compartir las diferentes prácticas y limitaciones que se pueden presentar en el sistema de salud colombiano al momento de tratar la PQRAD (tabla 3). El total de participantes manifestaron no tener conflictos de interés. Además, se contó con un panel revisor, quienes al ser profesionales reconocidos en el tema participaron en la revisión final del consenso.
Preguntas clínicas
Se contó con la participación de 10 especialistas en Nefrología, entre los cuales se asignó a una líder del consenso, quien realizó un listado de preguntas iniciales basadas en las necesidades prácticas para la atención de pacientes con PQRAD, las cuales fueron presentadas en su totalidad al panel de especialistas durante una sesión virtual y, por medio de la plataforma REDCap, los miembros del panel pudieron realizar una calificación referente a la pertinencia de cada pregunta, su apreciación respecto a la forma en cómo era formulada la pregunta y las anotaciones adicionales referentes a sugerencias. El total de los resultados fueron presentados en una sesión virtual, donde se realizó votación y según el 100% de acuerdo se definieron las preguntas a desarrollar. Se obtuvo un total de 18 preguntas a resolver de acuerdo con la experiencia profesional del panel evaluador (anexo A).
Tabla 3 Equipo desarrollador
| Papel en la elaboración de la guía | Participante y ciudad | Perfil profesional | Agremiación institucional |
| Líder | Angélica Roncallo Barranquilla | Médico/internista/ nefrólogo | Asociación Colombiana de Nefrología e Hipertensión Arterial (Asocolnef), Bogotá, Colombia |
| Experto clínico | Marcelo Aguirre Quibdó | Médico/internista/ nefrólogo/ epidemiólogo/ ultrasonido e intervencionismo/ máster en Patología Renal/Gerencia en Seguridad Social y Proyectos de Salud |
|
| Experto clínico | Luis Barros Barranquilla | Médico/internista/ nefrólogo |
|
| Experto clínico | Juan Carlos Conde-Manotas Barranquilla | Médico/internista/ nefrólogo/Cuidado Crítico |
|
| Experto clínico | Paula Gallón Blandón Medellín | Médico/internista/ nefrólogo |
|
| Experto clínico | Mauricio Lopera Medellín | Médico/internista/ nefrólogo |
|
| Experto clínico | José Gabriel López Medellín | Médico/internista/ nefrólogo |
|
| Experto clínico | Andrea Mantilla Popayán | Médico/internista/ nefrólogo clínico |
|
| Experto clínico | Theo Martínez Popayán | Médico/internista/ nefrólogo | Asociación Colombiana de Nefrología e Hipertensión Arterial (Asocolnef) |
| Experto clínico | Orlando Olivares Bogotá | Médico/internista/ nefrólogo | Asociación Colombiana de Nefrología e Hipertensión Arterial (Asocolnef) |
| Experto metodológico | Anderson Bermon Bucaramanga | Médico/ epidemiólogo/(c)PhD en Epidemiología y Bioestadística | Fundación Cardiovascular de Colombia (FCV) |
| Asesor externo | Cesar Augusto Restrepo Valencia Manizales | Médico/internista/ nefrólogo clínico e intervencionista |
|
| Asesor externo | Alfredo Osvaldo Wassermann Buenos Aires (Argentina) | Médico/especialista en Nefrología y Medio Interno/Diplomado en Gestión de Calidad |
|
Fuente: elaboración propia.
Búsqueda de la evidencia
Se realizó una búsqueda de literatura (anexo B) en las bases de datos PubMed, Scopus y Ovid, utilizando palabras clave como: "Polycystic Kidney, Autosomal Dominant"[Mesh] y utilizando los siguientes filtros: Full text, Clinical Study, Clinical Trial, Clinical Trial, Phase IV, Meta-Analysis, Practice Guideline, Pragmatic Clinical Trial, Randomized Controlled Trial, Systematic Review, in the last 10 years, Humans, English, French, Portuguese, Spanish.
Además, para Scopus se utilizó la siguiente consulta: ( TITLE ( adpkd ) OR TITLE ( "Autosomal Dominant Polycystic Kidney Disease") ) AND ( TITLE-ABS-KEY ( "Consensus") OR TITLE-ABS-KEY ( "Practice Guideline") OR TITLE-ABS-KEY ( "Controlled Trial") OR TITLE-ABS-KEY ( "Clinical trial") OR TITLE-ABS-KEY ( "Meta-Analysis") OR TITLE-ABS-KEY ( "Pragmatic Clinical Trial") ) AND PUBYEAR >2012 AND PUBYEAR <2024 AND ( LIMIT-TO ( SUBJAREA , "MEDI") ) AND ( LIMIT-TO ( DOCTYPE , "ar") OR LIMIT-TO ( DOCTYPE , "re") ) AND ( LIMIT-TO ( LANGUAGE , "English") OR LIMIT-TO ( LANGUAGE , "Spanish") OR LIMIT-TO ( LANGUAGE , "Portuguese") OR LIMIT-TO ( LANGUAGE , "French") ) AND ( LIMIT- TO ( EXACTKEYWORD , "Adult") ) y para Ovid "Autosomal Dominant Polycystic Kidney Disease".ti. and (Consensus or Practice Guideline or Clinical trial or Meta-Analysis).ab.).
Tamización y selección de la evidencia
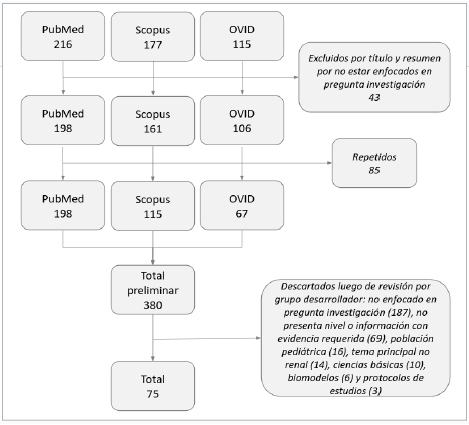
Un total de 508 artículos que fueron identificados y evaluados desde el título y el resumen de manera independiente por el grupo desarrollador, se descartaron inicialmente 43 por no estar enfocados en la pregunta de investigación. En una segunda valoración, realizada nuevamente por el grupo desarrollador, se tuvo en cuenta el total de documentos, se descartaron inicialmente 85 artículos por estar repetidos y 305 por cumplir criterios de exclusión los cuales eran no enfocarse en alguna de las preguntas de investigación, no presentar el nivel o la información con evidencia requerida, que la población de estudio fuera pediátrica, que el tema principal no fuera de implicaciones renales, que el enfoque del estudio fuera desde las ciencias básicas, y tuviera estudios con biomodelos y presentación de protocolos de estudios a realizar en un futuro. Finalmente, 75 artículos fueron los escogidos para la construcción del consenso (figura 2).
Los artículos revisados se presentan en el anexo C, discriminando su nivel de evidencia según la escala de Oxford [41].
Consenso formal de expertos
Las preguntas fueron distribuidas entre el total de miembros del grupo desarrollador, para realizar la revisión y generación de síntesis narrativas. Se realizaron videollamadas semanales para presentar estos textos y en construcción en conjunta se realizaban los cambios del caso.
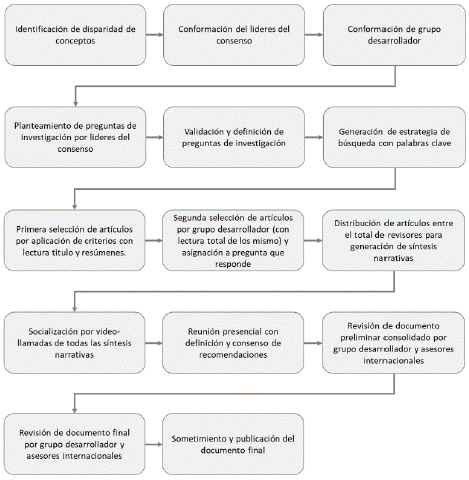
Una vez finalizadas las síntesis narrativas, se realizó una reunión presencial con el total de los miembros para realizar las recomendaciones para cada una de las preguntas. Una vez se contó con las recomendaciones, se realizó una primera versión del documento, la cual fue sometida para la evaluación de asesores externos, los cuales fueron escogidos por su experiencia y experticia en el tema, quienes manifestaron no tener conflictos de interés. Se suministró un enlace por REDCap para diligenciar la evaluación, que contó con una evaluación de pertinencia de las preguntas, valoración de la síntesis narrativa y porcentaje de acuerdo con la recomendación.
Se realizaron las correcciones finales adoptando las recomendaciones de los expertos y este documento final fue sometido a publicación (figura 3).
Graduación de las recomendaciones
Se realizó una reunión presencial tomando como parámetro la metodología Delphi, donde cada miembro del grupo desarrollador manifestó su nivel de acuerdo con cada una de las recomendaciones, siendo de 100 % el acuerdo entre todos los especialistas al finalizar las discusiones. Además, las recomendaciones contaron con soporte bibliográfico de artículos con el mayor nivel de evidencia encontrado y se categorizaron según el grado de recomendación de las guías del NHS (anexo C).
Resultados
Pregunta 1. ¿Cómo se diagnóstica la PQRAD?
La primera presunción diagnóstica debe guiarse por la historia familiar de primer grado de consanguinidad. El diagnóstico de los casos esporádicos, que representan el 10 % de los pacientes, se basa en los hallazgos imagenológicos y las características clínicas de la enfermedad, pero a veces es preciso un estudio genético, sobre todo en las fases iniciales de la enfermedad [4]. En algunos casos, el diagnóstico de la PQRAD debe fundarse en genética y en ayudas imagenológicas.
Estudio genético
Genéticamente la PQRAD es heterogénea, con dos genes identificados: el PKD1 y el PKD2 y se sabe que los efectos modificadores de los genes contribuyen a la alta variabilidad fenotípica de la PQRAD [12].
El análisis de ligamientos de ADN busca identificar la presencia de un segmento del cromosoma en el locus PKD1 o PKD2 que se segrega completamente con la enfermedad. Por lo tanto, no hay necesidad de identificar la mutación PQRAD exacta, ya que se está rastreando la presencia de estos marcadores y no las mutaciones en sí. La detección de mutaciones basadas en genes es el método más utilizado para el diagnóstico genético de PQRAD. Así, este enfoque busca identificar la mutación exacta en los genes PKD1 y PKD2 [42]. Se estima que aproximadamente el 10% de los pacientes no tienen una mutación identificable, por lo que estos pacientes suelen tener un fenotipo menor de PQRAD [43].
Aunque las pruebas genéticas para mutaciones de PQRAD están indicadas en algunos casos, estas no deben realizarse a todos los pacientes; y cuando no son necesarias, se puede hacer un diagnóstico certero con imágenes, manifestaciones clínicas y antecedentes familiares. Por otra parte, sí se debe considerar la realización de pruebas genéticas en posibles donantes vivos emparentados, para confirmar la ausencia de cualquier mutación para la PQRAD, lo mismo en pacientes sin antecedentes familiares de PQRAD (si la presentación imagenológica y síntomas extrarrenales son atípicos, la enfermedad renal es leve o se requiere información pronóstica), en familias con patrones imagenológicos atípicos de quistes renales, en familias afectadas por enfermedad poliquística de inicio temprano y en pacientes que desean un diagnóstico prenatal o preimplantacional [43,44].
La opinión de los expertos sugiere que el agrandamiento renal bilateral con innumerables quistes renales (>10 quistes por riñón) proporciona un diagnóstico "probable de PQRAD" y, en estas situaciones, las pruebas genéticas moleculares serían prudentes para confirmar el diagnóstico. A medida que la detección genética esté cada vez más disponible, es probable que las indicaciones para la prueba sean más inclusivas, ya que estos resultados enriquecen el pronóstico en la PQRAD. Hasta que las pruebas genéticas sean universalmente accesibles, se considerarán las siguientes indicaciones:
Confirmar el diagnóstico de PQRAD en el contexto de antecedentes familiares negativos.
Determinar el diagnóstico si las manifestaciones extrarrenales son sugestivas de síndromes distintos a la PQRAD o si la carga quística no es congruente con la función renal.
Excluir la PQRAD en donantes de riñón potenciales jóvenes que están en riesgo de PQ- RAD.
Confirmar el diagnóstico y descartar ciliopatías en el contexto de aparición temprana o muy temprana de la enfermedad [12].
Imagenología renal
Varias técnicas de imagen están actualmente disponibles para el diagnóstico y la evaluación de la progresión de PQRAD. Actualmente, la ultrasonografía renal es el método de diagnóstico electivo inicial de tamizaje o incidental para detectar y diagnosticar la enfermedad en pacientes con un familiar afectado, ya que es un enfoque práctico y rentable, sin embargo, existe la preocupación que, en comparación con la tomografía computarizada (TC) o la resonancia magnética nuclear (RMN), la ultrasonografía depende del operador y tiene una mayor variabilidad, lo que dificulta la obtención de resultados precisos y reproducibles. Así, la RMN es la mejor técnica de imagen para definir el volumen renal total (VRT), especialmente de forma repetida, al no exponer al paciente a radiación. Para la determinación del VRT ajustado por altura, la estereología por RMN se considera la modalidad de imagen preferida, siendo incluso el estándar de oro por tener mayor exactitud y precisión en comparación con las otras técnicas. En caso de no contar con la estereología por RMN, se suele hacer el cálculo del VRT ajustado por la altura, mediante la fórmula de planimetría por RMN o ecuación de elipsoide por RMN. El cálculo del VRT por ecuación de elipsoide también se ha validado con estudios de imagen por TC. Recientemente, se ha informado que los métodos automatizados de determinación de VRT producen resultados similares a la estereología; para estas medidas, los pacientes se clasifican en función del VRT ajustada por altura y la edad [4,44].
Las nuevas técnicas ecográficas, incluida la ultrasonografía tridimensional y de alta resolución, tienen algunos datos en el diagnóstico de PQRAD, pero estas modalidades no están ampliamente disponibles y su desempeño en la determinación del tamaño no ha sido evaluado [44].
Criterios clínicos (síntomas y antecedentes familiares)
Es una enfermedad sistémica caracterizada por el desarrollo temprano de quistes renales que crecen inexorablemente con el tiempo, lo que lleva a la destrucción del parénquima renal y la pérdida de la función renal. La tabla 1 (manifestaciones clínicas) describe los criterios clínicos de la enfermedad.

Pregunta 2. ¿Cuáles son los criterios imagenológicos para el diagnóstico de PQRAD (por ultrasonografía, tomografía y resonancia)?
Las imágenes en PQRAD se pueden clasificar en dos categorías: imágenes renales para el diagnóstico e imágenes para el pronóstico y la progresión de la enfermedad. El método preferido para confirmar la presencia de PQRAD en pacientes con antecedentes familiares es la ultrasonografía y el uso de los criterios unificados (el cual discrimina según genotipo PKD1, PKD2 o para genotipo desconocido y según rango de edad) (tabla 4). En ausencia de antecedentes familiares, no existen criterios validados para diagnosticar PQRAD basado en imágenes de ultrasonografía renal. En circunstancias seleccionadas, como por ejemplo la falta de antecedentes familiares y pacientes jóvenes, el diagnóstico de PQRAD puede requerir imágenes de mayor sensibilidad, además de la evaluación de manifestaciones extrarrenales y pruebas genéticas. Además, existen criterios descritos por RMN que permiten el diagnóstico específico a una edad temprana y son especialmente útiles cuando los resultados de la ecografía son equívocos [45,46] (tabla 5).
La sensibilidad de la ultrasonografía clásica para pacientes con PKD1 es significativamente mayor que en los pacientes con PKD2.

Tabla 4 Criterios unificados de ultrasonografía para el diagnóstico de PQRAD en personas con un riesgo del 50 % de padecer PQRAD según los antecedentes familiares
| Edad en años | PKD 1 | PKD2 | Tipo de gen PQRAD desconocido |
|---|---|---|---|
| 15-29 |
≥ 3 quistes" VPP: 100 % SEN: 94,3 % |
≥ 3 quistes" VPP: 100 % SEN: 69,5 % |
≥ 3 quistes VPP: 100 % SEN: 81,7 % |
| 30-39 |
≥ 3 quistes" VPP: 100 % SEN: 96,6 % |
≥ 3 quistes" VPP: 100 % SEN:94,9 % |
≥ 3 quistes" VPP: 100 % SEN: 95,5 % |
| 40-59 |
≥ 2 quistes en cada riñón VPP: 100 % |
≥ 2 quistes en cada riñón VPP: 100 % |
≥ 2 quistes en cada riñón VPP: 100 % SEN: 90,0 % |
| SEN: 92,6 % | SEN: 88,8 % | ||
Nota: pacientes ≥ 60 años, con ≥ 4 quistes en cada riñón es un criterio diagnóstico; además, todos los valores son presentados en promedios; PPV: valor predictivo positivo; SEN: sensibilidad. "Unilateral o bilateral
Fuente: [46].
Tabla 5 Criterios de resonancia magnética para el diagnóstico de PQRAD en personas con un riesgo del 50 % de padecer PQRAD según los antecedentes familiares
| Criterios diagnósticos por resonancia magnética para PQRAD | |
|---|---|
| Sujetos entre 16 y 40 años con riesgo de presentar PQRAD: |
• >10 quistes entre los dos ríñones: PQRAD • <10 quistes entre los dos riñones: no PQRAD |
| Sujetos entre 16 y 40 años con riesgo de presentar PQRAD y que deseen ser donantes renales | • <5 quistes entre los dos riñones: aceptable como donante. |
| Ante un resultado no concluyente de las pruebas de imagen, se recomienda realizar un estudio genético. | |
Fuente: [45].
Pregunta 3. De acuerdo con el criterio imagenológico, ¿cómo se clasifica la PQRAD?
Las imágenes renales se pueden usar para caracterizar la PQRAD como una presentación típica o atípica usando la clasificación de la Mayo Clinic (tabla 6). De acuerdo con este sistema, los pacientes con una típica distribución simétrica, bilateral y difusa de los quistes se clasifican como clase 1 (aproximadamente el 90 % de los pacientes) y los pacientes con distribución atípica, asimétrica o segmentaria de los quistes, que se clasifican como clase 2 [43] (tabla 6).
Tabla 6 Clasificación de la PQRAD basada en caracterización imagenológica de acuerdo con la clasificación de Mayo Clinic
| Clase, subclase y término | Descripción |
|---|---|
| 1. PQRAD típica | Distribución bilateral y difusa, con reemplazo leve, moderado o severo del tejido renal por quistes, donde todos los quistes contribuyen de manera similar al VRT |
| 2. PQRAD atípica | |
| Unilateral | Afectación quística difusa de un riñón que causa agrandamiento renal marcado con un riñón contralateral normal, definido por un volumen renal normal (<275 ml en hombres; <244 ml en mujeres) y tener 0-2 quistes. |
| Segmentaria | Enfermedad quística que afecta solo a un polo de uno o ambos riñones y que respeta el tejido renal restante. |
| Asimétrica | Compromiso quístico difuso de un riñón que causa agrandamiento renal marcado con compromiso difuso segmentario leve o mínimo del riñón contralateral, definido por una pequeña cantidad de quistes (>2 pero <10) y volumen que representa <30 % de VRT. |
| Desequilibrada | Distribución bilateral de quistes renales con reemplazo leve de tejido renal con quistes atípicos donde ≤ 5 quistes representan ≥ 50 % VRT (el diámetro de quiste más grande se usa para estimar el volumen de quiste individual). |
| Presentación bilateral con atrofia unilateral adquirida | Compromiso quístico difuso de un riñón que causa agrandamiento renal de moderado a severo con atrofia adquirida contralateral. |
| Presentación bilateral con atrofia bilateral | Deterioro de la función renal (creatinina sérica ≥ 1,5 mg/dl) sin agrandamiento significativo de los riñones, definido por una |
Fuente: [43].
La RMN o la TC para calcular el VRT ajustado a la altura y la edad son actualmente los métodos recomendados para la evaluación del tamaño renal en pacientes con PQRAD.
Cuando las imágenes por RMN no están disponibles, las imágenes por TC se pueden usar para medir el VRT. Si no se dispone de imágenes por RMN y TC, el uso de la longitud renal medido por ultrasonografía renal es un sustituto que puede llegar a usarse.
El VRT ajustado por altura y edad se realiza para la evaluación inicial de pacientes con PQRAD, donde la imagen de RMN ponderada en T2 permite medir el VRT mediante la ecuación elipsoide basada en la longitud, el ancho y el grosor ortogonales del riñón. La ecuación del elipsoide es: para cada riñón = n x w x t/6, donde / = largo, t = espesor y w = ancho. La clasificación de imágenes de la Mayo Clinic facilita el uso de VRT [22]. El método del elipsoide estima el VRT y proporciona una herramienta práctica que le permite al nefrólogo medir el VRT en minutos.
Por otra parte, las imágenes abdominales generales mediante RMN son útiles para obtener imágenes de referencia del hígado, el bazo y el páncreas para controlar los cambios en los quistes hepáticos o el crecimiento de quistes pancreáticos o esplénicos [43].
Las imágenes renales, junto con la clasificación de la Mayo Clinic, se utiliza con fines pronósticos. Además, tienen implicaciones en la selección de pacientes que pueden beneficiarse del inicio de tratamiento farmacológico dirigido. Al integrar el VRT, ajustado para la altura más la edad, se pueden dividir los pacientes de clase 1 en las subclases de la A a la E, donde las clases 1C, 1D y 1E muestran la más alta tendencia a desarrollar enfermedad renal de aparición temprana; mientras que los pacientes de clase 2 generalmente no tienen riesgo de progresión rápida de la enfermedad. Una vez que los pacientes se clasifican de acuerdo con los criterios de la Mayo Clinic, se puede predecir la progresión de su enfermedad a lo largo del tiempo. Sólo los pacientes con presentación típica y edades entre 15 y 80 años aplican en el calculador de la Mayo Clinic para evaluar por RMN o TC el VRT ajustado a la altura y edad. Esta herramienta está disponible en el sitio educativo de la Clínica Mayo bajo el nombre "Imaging classification of ADPKD: A simple model for selecting patients for clinical trials", bajo el enlace https:/www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754, que fue consultado el 28 de noviembre del 2024. La herramienta también permite clasificar a los pacientes si previamente se calculó el VRT por estereología.

Pregunta 4. ¿Cuáles son las indicaciones para solicitar estudio genético en pacientes con PQRAD?
En la actualidad hay acceso a la realización del estudio genético para el diagnóstico de PQRAD, sin embargo, tiene un costo elevado, por lo que no está indicado si el diagnóstico clínico e imagenológico es claro.
El análisis mutacional requiere ser realizado en laboratorios de experiencia, ya que esta técnica tiene dificultades por la gran heterogeneidad de los alelos de los genes. En la actualidad, se conocen más de 100 genes vinculados a quistes renales.
El método más utilizado para el diagnóstico genético de PQRAD es la secuenciación clásica de Sanger o la secuenciación masiva, aunque tiene una limitación, ya que la sensibilidad es inferior al 100 %, por lo que no confirma y tampoco descarta la sospecha diagnóstica en caso de no tener el resultado mutacional [47,48].
La secuenciación clásica de Sanger es el análisis secuencial (no simultáneo) de los 46 exones y regiones flanqueantes de PKD1, que es el más frecuentemente mutado en la PQRAD y los 15 de PKD2 se realizan cuando algún familiar presenta ERC E5 con necesidad de diálisis después de los 70 años [49].
Si la secuenciación no identifica ninguna variante patogénica (truncante) o una variante claramente patogénica, debe realizarse la técnica MLPA (Multiplex Ligation-dependent Probe Amplification), ya que esta detecta deleciones y duplicaciones en un 4 % de los casos [50].
La secuenciación masiva permite la secuenciación de PKD1, PKD2, GANAB y DNAJB11 de forma simultánea con otros genes asociados a enfermedades renales quísticas, lo que genera información de fenotipos poco frecuentes y reduce el tiempo para el diagnóstico [51].
Por otra parte, existe el análisis mutacional de ligamiento en familias, actualmente utilizado únicamente en los casos familiares con clínica de PQRAD absolutamente certera, en los que en la secuencia de genes causantes de la PQRAD no se han identificado variantes patogénicas. Esta técnica requiere de la intervención de algunos miembros de la familia con diagnóstico de PQRAD y no se recomienda en pacientes sin antecedentes familiares, ni en pacientes con clínica dudosa de PQRAD [52, 53].
También existen indicaciones para la realización del estudio genético en pacientes con:
HTA previa a los 35 años o presencia de síntomas urológicos, con el objetivo de determinar el riesgo de rápida progresión y justificar el tratamiento específico. En los pacientes con presencia de mutación en PKD1, se puede aplicar el PROPKD score de riesgo (ver más adelante).
En familiares de pacientes con PQRAD y que son candidatos a donante vivo con ultrasonografía no concluyente, diagnóstico de PQRAD por imagen atípico como poliquistosis renal asimétrica e insuficiencia renal sin significativo agrandamiento renal, gravedad de la enfermedad muy discordante entre miembros de una misma familia y diagnóstico de PQRAD leve o de inicio temprano, muy grave o con características sindrómicas [47].
Pacientes que desean asesoría genética preconcepcional.
En personas con presentación típica de PQRAD, pero con un familiar con presentación muy precoz. En estos casos el estudio genético puede identificar un alelo hipomórfico además del alelo con la mutación patogénica o un alelo hipomórfico en ambas copias de PKD1 [47].
En pacientes sin antecedentes familiares de PQRAD y en los que no se han identificado mutaciones en el gen PKHD1 (causante de la poliquistosis renal autosómica recesiva) o con características imagenológicas de PQRAD.
Ante un resultado no concluyente de las pruebas de imagen y en pacientes sin antecedentes familiares de PQRAD jóvenes o con diagnóstico clínico incierto.
Familias con múltiples sujetos con quistes renales con patrón imagenológico atípico de PQRAD, candidatos a un diagnóstico diferencial de otras enfermedades renales quísticas[4].
El diagnóstico genético para identificar el gen causante de la enfermedad es cuestionado por la variabilidad clínica que genera cada mutación asociada al gen, no obstante, se describen asociaciones entre genotipo-fenotipo y su peso en la decisión de inicio de tratamiento [54].

Pregunta 5. ¿Cuáles son los diagnósticos diferenciales de la PQRAD?
Las personas con PQRAD con frecuencia tienen antecedentes familiares de dicha enfermedad, sin embargo, existen pacientes sin antecedentes familiares, lo cual requiere un cuidadoso trabajo diagnóstico para diferenciar entre PQRAD y otras enfermedades renales quísticas [55].
Presentamos la tabla 7 con las condiciones para diagnóstico diferencial, con sus características genéticas y clínicas relevantes.
Pregunta 6. ¿Cómo evaluar la progresión de la enfermedad PQRAD?
Al considerar la característica multifactorial de las condiciones de riesgo de susceptibilidad, inicio y progresión de la enfermedad renal crónica, sin especificar su etiología individual, se debe entender que no existen herramientas de uso clínico o estudios paraclínicos que permitan desagregar claramente la progresión de la PQRAD per se, versus la aparición, el desarrollo y la progresión de la comorbilidad o las complicaciones cerebrocardiovasculares en cada caso individual (figura 4).
Tabla 7 Condiciones para un diagnóstico diferencial
| Condición / trastorno / enfermedad | Gen | Herencia | Historia familiar | Prevalencia | Características clínicas por compromiso renal / extrarrenal |
|---|---|---|---|---|---|
| Quistes renales simples benignos múltiples | N/A | Adquirida | Ninguna | Común | Relativamente común en población general; aumento en número y tamaño con la edad (<30 años: poco común, raramente bilateral; 30-59 años: poco común tener al menos 2 quistes en cada riñón; >60 años: poco común, tener >4 quistes en cada riñón). |
| Enfermedad renal quística adquirida | N/A | Adquirida | Ninguna | Común | Hay ERC avanzada, particularmente en pacientes con diálisis; los quistes son pequeños, bilaterales y múltiples (>4 quistes en cada riñón); los riñones son entre pequeños y normales. |
| Riñón en esponja medular (Lenarduzzi-Cacchi- Ricci disease) | Desconocido | Poco claro; autosómico dominante en algunos casos | Agrupación familiar | 1 en 5000 | Malformación de los túbulos colectores distales, deterioro de la función renal, acidosis tubular, infecciones recurrentes del tracto urinario. Nefrocalcinosis medular, numerosos quistes diminutos dentro de la médula renal y compromiso bilateral de las lesiones. |
| Complejo esclerosis tuberosa | TSC 1-2 | Autosómica dominante | Ausente de 2/3 casos | 1 en 10 000 |
La deleción contigua de PKD1/TSC2 da como resultado poliquistosis grave de inicio temprano con ERC E5 durante las primeras dos décadas de vida. Lesiones cutáneas (angiofibromas faciales, fibroma periungueal, máculas hipomelanóticas y parche de Shagreen); hamartomas en piel, SNC, pulmón, hueso, retina; tubérculo cortical; astrocitoma subependimario de células gigantes; rabdomioma cardiaco; linfangioleiomiomatosis pulmonar. Numerosos quistes renales bilaterales (14-32 %). Triada de síntomas clínicos clásicos: angiofibroma facial, retraso mental y epilepsia. |
| Nefronoptisis | NPHP1-6, DZIP1L, NPHP1-20, NPHP1L, NPHP2L, TRAF31P1, AH11, CC2D2A | Autosómica recesiva | 1 en 10 000 - 1 en 50 000 | Riñones de tamaño normal con quistes en la unión corticomedular; nefritis tubulointersticial crónica; trastorno en la concentración urinaria. | |
| Enfermedad renal Poliquística autosómica recesiva | PKDH1 | Autosómica recesiva | 25 % concordancia entre hermanos | 1 en 20000 | Riñón temprano en la vida, quístico, aumentado de tamaño y ecogénico. Con la edad, los riñones son más pequeños, con quistes macroscópicos, nefrocalcinosis o pequeñas calcificaciones medulares comunes; oligohidramnios (fenotipo de Potter) e hipoplasia pulmonar intrauterina, siempre fibrosis hepática congénita y enfermedad de Caroli. |
| Síndrome Von Hippel-Lindau | HVL | Autosómica dominante | ∼20 % de novo | 1 en 5000 - 1 en 36 000 | Numerosos quistes de tamaño variado en más del 50 % de los pacientes (59-63 %), acompañados de lesiones sólidas. Hemangioblastomas cerebeloso y espinal, de retina, quistes pancreáticos, adenomas quísticos, tumores neuroendocrinos pancreáticos, feocromocitoma, cistoadenomas epididimarios. |
| Síndrome Bardet- Biedl (es una variante de la nefronoptis) | BBS 1-12 | Autosómica recesiva | 1 en 140 000 | Degeneración retiniana, obesidad infantil, retraso mental, malformaciones del tracto urogenital y polidactilia. | |
| Síndrome orofaciodigital tipo 1 | OFD1 | Dominante ligada a X | 1 en 250 000 | Varón embrionario letal, paladar hendido, lengua bífida, frenillo hiperplásico, hipertelorismo, cresta nasal ensanchada, anomalías digitales que incluyen sindactilia, malformaciones del SNC. Solo afecta a mujeres. | |
| Enfermedad renal quística medular | MCKD1-2 | Autosómica dominante | Rara | Desconocida | Fibrosis intersticial. Numerosos quistes en la interfaz entre la médula renal y la corteza, y dentro de la médula renal. |
| Quistes renales y síndrome de diabetes (RCAD/MODY5/H NF-1Bb) | HNF1B | Autosómica dominante | Mutaciones espontáneas (a menudo deleciones) en un 50 % de los casos | Desconocida | Quistes o malformaciones renales en el 90 %, diabetes mellitus en el 45 % de los casos, hipomagnesemia en el 40 %, anomalías del tracto genital en el 20 %, hiperuricemia en el 20 % y enzimas hepáticas elevadas en un 15 %. |
| Riñón displásico multiquístico | N/A | N/A | N/A | 1/1000 - 1/4000 | Unos pocos (es decir, carcinoma quístico de células renales, nefroma quístico, nefroblastoma quístico parcialmente diferenciado, tumor mixto epitelial y estromal). |
| Nefropatía tubulointersticial autosómica dominante por mutaciones en genes UMOD/MUC1 | UMOD/ MUC1 | Autosómica dominante | N/A | Sin datos | Nefropatía tubulointersticial, gota frecuente o hiperuricemia, a veces quistes en la unión cortico- medular. |
| Nefropatía Tubulointersticial autosómica dominante por mutaciones en gen REN | REN | Autosómica dominante | N/A | Sin datos | Hipotensión, hiperuricemia e hiperpotasemia. |
| Enfermedad renal en tubulointersticial y autosómica dominante | UMOD, MUC1, REN, HNF1B, SEC61A, DNAJB11 | Autosómica dominante | N/A | Desconocida |
Fibrogénesis tubulointersticial y deterioro lento progresivo de la función renal. Pequeños quistes renales sin agrandamiento del riñón. Anomalías circulatorias tipo AIC, agrandamiento de la aorta torácica y disociación de la arteria carótida. |
| Síndrome Meckel- Gruber | MGS1-6 | Autosómica recesiva | 1/13 250 - 1/140 000 | Encefalocele occipital y polidactilia postaxial | |
| Poliquistosis hepática autosómica dominante | PRKCSH, SEC63, LRP5 ADPLD- GANAB, ALG8 y SEC61B | Autosómica dominante | Poliquistosis renal autosómica dominante, enfermedad renal crónica y poliquistosis hepática. |
Nota: ERC: enfermedad renal crónica; ERC E5: enfermedad renal crónica estadio 5; N/A: no aplica; SNC: Sistema nervioso central.


Fuente: elaboración propia.
Fuente: elaborado en consenso por los autores con base en [56].
Figura 4 Factores relacionados con la progresión de la PQRAD
Al identificar estos factores se da por entendida la pertinencia de solicitar estudios paraclínicos por imagen, biomarcadores séricos y urinarios, copeptina, análisis en orina casual o de 24 horas para albuminuria y proteinuria [56].
Progresión de la PQRAD
La tasa de filtración glomerular permanece normal durante años debido a la filtración glomerular adaptativa en las nefronas restantes [57], por lo que esta medida al inicio de la enfermedad no es un buen marcador de progresión [43].
Las mutaciones de PKD1 (particularmente truncada), hipertensión arterial, proteinuria y el VRT ajustado por la altura y edad se han identificado como predictores principales de la progresión de la PQRAD [58].
Un estudio diseñado en Francia creó un modelo predictivo basado en genética para predecir la progresión de los pacientes con PQRAD, llamado Score PRO-PKD. En él se incluye la presencia de la mutación PKD1 truncada (4 puntos), el género (masculino 1 punto), la aparición de la hipertensión arterial antes de los 30 años (2 puntos) y las complicaciones urológicas antes de los 35 años (hematuria macroscópica, dolor en flanco e infección de quistes) (2 puntos). Los pacientes con un score mayor a 6 puntos experimentan una rápida progresión y deben ser tratados. Con un score menor de 3 hay una lenta progresión y no se requiere tratamiento. Si el score está entre 3 y 6, el pronóstico no es claro [54,59].
Al referirnos a los factores genéticos implicados en la progresión de la PQRAD, se debe considerar: heterogeneidad de locus y heterogeneidad alélica: tipo de mutación y localización, alelos hipomórficos y genes modificadores que condicionan una forma más grave de PQRAD [53,56,60-64].
Se han identificado nuevos biomarcadores pronósticos que varían desde la evaluación de la gravedad de la enfermedad basada en la capacidad de concentración de orina en pacientes después de la privación de agua, hasta la observación de proteínas séricas y urinarias, péptidos y metabolitos [65-67]. Está claro que los biomarcadores de líquidos (suero y orina) pueden ser prometedores desde una perspectiva de respuesta pronóstica y potencialmente terapéutica [68,69].
Otros factores pronósticos
En el estudio CRISP (Consortium for radiologic imaging studies of polycystic kidney disease), los niveles más altos de actividad de la vasopresina (medida utilizando la osmolalidad de la orina de 24 horas como marcador sustituto) estuvieron asociados con una mayor disminución en la TFG del año 1 al 6 [43,70].
Por otro lado, los niveles elevados de ácido úrico en suero se asociaron con una progresión de la enfermedad y se ha demostrado un aumento del 5,8 % en el VRT y un aumento del 4,1 % en la relación VRT/área de superficie corporal por cada aumento de 1 mg/dl (59,5 ^mol/l) de ácido úrico [43, 71], aunque esta asociación se ve comprometida al realizar ajuste al análisis de manera independiente de este biomarcador [72].
Para definir adecuadamente una rápida progresión, es necesario comprender la prolongada historia natural de la enfermedad. En la PQRAD, la función renal permanece normal durante las primeras décadas de la vida, a pesar del crecimiento del VRT, por lo cual cobra gran utilidad la valoración de este para determinar el pronóstico [4,73].
Las recomendaciones de la Agencia Europea de Medicamentos (EMA, según sus siglas en inglés) no especifican qué se entiende por rápida progresión. El Working Group of Inherited Kidney Diseases (WGIKD) de la European Dialysis and Transplant Association (EDTA) junto al grupo de European Renal Best Practice Guidelines (ERBP) propusieron una definición de rápida progresión en la PQRAD que ha quedado actualmente obsoleta [4].
Respecto a la valoración de la rápida progresión de la enfermedad renal, desde el punto de vista de la TFG-e, el consenso de KDIGO la define como la pérdida de más de 5 ml/min/1,73m2/año de TFG-e, pero esta definición parece demasiado estricta para pacientes con PQRAD [4,74].
El VRT es el mejor predictor de la progresión de la ERC [4, 73, 75] y se afirma que los pacientes que muestran un aumento anual >5 % en VRT deben clasificarse en mayor riesgo de progresión de la enfermedad [76, 77]. Conforme aumenta el volumen renal, mayor es la velocidad de descenso de la TFG-e [4, 73].
Hay estudios que han validado los modelos de predicción de la Mayo Clinic para PQRAD, entre ellos, un estudio suizo concluyó que los modelos de predicción de la Mayo Clinic se pueden generalizar a otros entornos clínicos y proporcionan una herramienta precisa basada en predictores disponibles para identificar a los pacientes con alto riesgo de progresión rápida de la enfermedad, con las categorías que surgen del VRT ajustado a altura y edad [78].
Las guías canadienses recomiendan que en la práctica clínica actual, los pacientes con medición de VRT ajustado a altura se clasifiquen en términos de su riesgo de progresión, según la clasificación de la Mayo Clinic y otras herramientas clínicas validadas [43].
Por otro lado, el grupo de trabajo de la Asociación Renal Europea (ERA, por sus siglas en inglés) sobre trastornos renales hereditarios, la Red Europea de Referencia de Enfermedades Renales Raras y la Asociación Internacional de Enfermedades Renales Poliquísticas recomiendan que ante una disminución anual confirmada de la TFG-e ≥ 3 ml/min/1,73m2 se define una rápida progresión de la enfermedad. La estimación de la pérdida de TFG-e debe ser fiable y basarse en al menos cinco mediciones durante un periodo ≥ 4 años (considerando la etapa de hiperfiltración y la disminución abrupta una vez pasada esta etapa) y recomiendan que se evalúen y excluyan otras causas de disminución de la TFG-e como factores contribuyentes importantes, especialmente en casos de disminución no lineal de la TFG-e, en pacientes mayores o pacientes con múltiples comorbilidades que pueden tener un impacto en la TFG-e. Este grupo utiliza la clasificación de la Mayo Clinic como método primario para la predicción del riesgo, donde los pacientes de clase 1C deben considerarse cuidadosamente, debido a la superposición con la enfermedad de progresión lenta y se debe buscar evidencia adicional de progresión rápida de la enfermedad en estos pacientes [76].
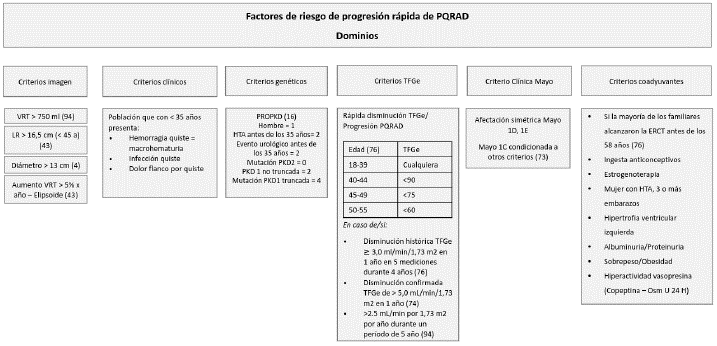
Por otro lado, diferentes sociedades internacionales han planteado los criterios de rápida progresión y establecen sus guías de manejo y recomendaciones para el uso de tolvaptán, en la tabla 8 se encuentra los puntos relevantes que toca cada guía. En la figura 5 se describen por dominios de los factores implicados y descritos para validar progresión en PQRAD.

Pregunta 7. Herramientas bioquímicas e imagenológicas validadas en el seguimiento del paciente con PQRAD
Para el seguimiento de la PQRAD los estudios imagenológicos son una herramienta útil para establecer pronóstico, progresión del compromiso renal y detectar complicaciones extrarrenales en el paciente [3]. El VRT es una medida validada para evaluar la progresión de la enfermedad renal y reconocida como un biomarcador pronóstico, aprobado por la Administración de Drogas y Alimentos de Estados Unidos (FDA, según sus siglas en inglés) y la EMA [3].
Tabla 8 Comparación entre guías y consensos
| Criterios | España, 2020 | Estados Unidos, 2018 | Canadá, 2018 | ERA-EDTA, 2021 | Japón, 2014 | China, 2020 | |
|---|---|---|---|---|---|---|---|
| Imagen | RMN o TC para medir VRT ajustado a altura y edad | Lo incluye | Lo incluye | Lo incluye | Lo incluye | Lo incluye | Lo incluye |
| MAYO 1C, 1D, 1E | Rápida progresión al 1D y 1E, y para 1C asociarse a otro criterio | Lo incluye | Lo incluye | Clase 1C de la Mayo Clinic debe considerarse cuidadosa mente | N/A | Lo incluye | |
| Aumento de VRT >5 % por año | N/A | N/A | Lo incluye + VRT de 750 ml | N/A | VRT ≥ 750ml | N/A | |
| Longitud del riñón evaluada por ultrasonografía renal > 16,5 cm en pacientes < 45 años | >13 cm por ultrasonografía y valorar en función de la edad | N/A | Lo incluye | Lo incluye | N/A | N/A | |
| Genéticos | La puntuación PROPKD | Lo incluye | N/A | N/A | Lo incluye | N/A | N/A |
| >35 años sin complicacion es o con información clínica faltante, Adicionar estudio genético (valor pronóstico de mutación truncada PKD1) | N/A | N/A | N/A | Lo incluye | N/A | N/A | |
| TFG-e | Declive de TFG-e 5 ml/min/1,73 m2/ año o >2,5 ml/min/1,73 m2/año por 5 años. | VRT ajustado por altura y edad (clasificación de Mayo Clinic 1C: si el declive de TFGe de los últimos 3 años es ≥ 3,5 ml/año) | N/A | Declive de TFG-R > 2,5 ml/min/1,7 3m2/año. | Declive anual TFGe ≥ 3 ml/min/1,73 m2/año en 5 mediciones, periodo de más de 4 años | N/A | N/A |
Fuente: elaboración propia.
No es necesario realizar estudios de TC y RMN de forma seriada o de rutina, o repetir las mediciones del VRT después de una prueba inicial, sino que debe haber un intervalo de al menos un año entre pruebas o idealmente más tiempo [3,76]. Repetir imágenes en menor tiempo de seguimiento estaría recomendado cuando exista alguna circunstancia que modifique la enfermedad o ante la sospecha de otra patología como litiasis o tumores [4,22].
Otros parámetros de seguimiento
Por lo general, la progresión de la PQRAD se controla a través de cambios en los niveles de creatinina sérica y la estimación de la tasa de filtrado glomerular (TFG), sin embargo, estas medidas proporcionan información limitada [11]. En cambio, se debe estimar la TFGe utilizando la ecuación CKD-EPI (creada por el grupo de colaboración de epidemiología de insuficiencia renal crónica, Chronic Kidney Disease Epidemiology) para monitorear la función renal en pacientes con PQRAD [22,79].
Proteinuria y albuminuria: deben medirse para controlar la progresión de la PQ-RAD [22, 79]. Aproximadamente el 25 % de los pacientes diagnosticados con PQRAD tienen proteinuria (>300 mg/día, pero generalmente no excede 1 g/día). La proteinuria se asocia con una disminución más rápida de TFGe y al desarrollo de ERC en menor tiempo; en un análisis post hoc del ensayo TEMPO 3, la presencia de un grado mayor de albuminuria en estadios tempranos se asoció a una mayor pérdida de TFG [76, 80]. Por otro lado, la proteinuria en rango nefrótico puede orientar sobre la coexistencia de otro trastorno renal [22].
La tabla 9 resume las pruebas de laboratorio sugeridas para pacientes con PQRAD en los diferentes estadios de la ERC.

Tabla 9 Pruebas de laboratorio sugeridas para pacientes con PQRAD
| Laboratorio | Estadio 3a | Estadio 3b | Estadio 4 | Estadio 5* |
|---|---|---|---|---|
| Creatinina, BUN, electrolitos | Cada 6 meses | Cada 3 meses | Cada 2 meses | Mensualmente |
| Albumina, calcio, fósforo | Cada 6 meses | Cada 3 meses | Cada 3 meses | Cada 3 meses |
| AST, ALT, GGT, bilirrubinas** | Cada 6 meses | Cada 6 meses | Cada 4 meses | Cada 3 meses |
| Relación albuminuria/ creatinuria (RAC) | Cada 6 meses | Cada 6 meses | Cada 6 meses | Cada 6 meses |
| Ácido úrico | Cada 6 meses | Cada 6 meses | Cada 4 meses | Cada 3 meses |
Nota: *Paciente con manejo médico, sin diálisis y **Cuando exista compromiso hepático; PTHi: Paratohormona intacta; AST: Aspartato aminotransferasa; ALT: Alanina aminotransferasa; GGT: Gamma-glutamil transferasa.
Fuente: adaptado de [3].
Pregunta 8. ¿Cuáles son las intervenciones generales y farmacológicas en el manejo de la PQRAD? (hipertensión, proteinuria, entre otros)
Intervenciones generales y farmacológicas
Recientemente, una mayor comprensión de la fisiopatología de la PQRAD y los avances genéticos han llevado a nuevos enfoques de tratamiento, dirigidos a las vías fisiológicas, que se ha demostrado que retrasan la progresión de la enfermedad.
Se puede utilizar con éxito un enfoque múltiple con tratamientos farmacológicos y no farmacológicos para disminuir la tasa de progresión de la PQRAD hacia la insuficiencia renal.
Las medidas de apoyo tienen como objetivo reducir la morbilidad y la mortalidad asociadas con las manifestaciones de la PQRAD.
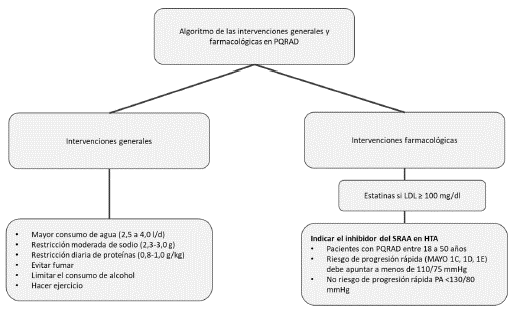
Estos incluyen un control estricto de la presión arterial <110/75 mmHg en jóvenes, <130/80 mmHg en mayores de 50 años y un mayor consumo de agua, lo que puede disminuir los niveles de vasopresina, ya que está descrito que la secreción de vasopresina está controlada principalmente por la osmolaridad sérica y, en consecuencia, la ingesta de agua [81].
Estudios en humanos no han demostrado que el consumo de agua afecte la progresión de la enfermedad en PQRAD, sin embargo, el consumo de agua suprime la hormona antidiurética (ADH), lo que resulta en atenuación en el crecimiento de los quistes y proliferación de la célula quística, por lo que se recomienda una ingesta de 2,0 a 3,0 litros de agua al día, que ha demostrado conseguir una osmolalidad urinaria <280 mOsm/kg en la primera orina de la mañana. Esta recomendación debe ser tomada con cautela cuando la enfermedad renal está muy avanzada [22, 77, 82].
Una restricción moderada de sodio en la dieta (2,3-3,0 g) y una ingesta diaria de proteínas en la dieta de 0,8-1,0 g/kg de peso corporal es ideal para retrasar la progresión de la enfermedad renal crónica [12, 83]. En los pacientes con PQRAD, la elevada ingesta de proteínas induce la hiperfiltración y el incremento de los niveles de vasopresina, lo cual lleva al aumento de tamaño de los quistes y la declinación de la función renal. Aun cuando no existe evidencia clara de un efecto beneficioso de la restricción en la ingesta proteica en la PQRAD, parece aconsejable consumir alrededor de 0,8 mg de proteínas/kg de peso/día [82,84].
Además, los pacientes con PQRAD deben evitar fumar y limitar el consumo de alcohol e incorporar la actividad física y el ejercicio dentro de las pautas de manejo [81,85,86].
Estatinas
Se conocen efectos antiproliferativos, antiinflamatorios y antioxidantes de los inhibidores de la HMG-CoA (3-hidroxi-3-metilglutaril-CoA) reductasa. Estos efectos se han demostrado independientemente de la reducción del colesterol [70,87].
También se evidenció que los niveles basales de colesterol HDL en suero tenían un efecto protector contra el aumento del crecimiento renal y disminuciones más rápidas en la TFG [70, 88].
Debido a que las guías KDIGO (Kidney Disease: Improving Global Outcomes) recomiendan el uso de estatinas para la protección cardiovascular en todos los pacientes con ERC que tienen más de 50 años y no reciben diálisis, estos medicamentos pueden desempeñar un papel secundario en pacientes con ERC con PQRAD, para ayudar a prevenir el crecimiento de quistes [74, 81].
Hipertensión
Una de las complicaciones más comunes de la PQRAD es el desarrollo de hipertensión. Los pacientes hipertensos muestran una mayor activación del SRAA, en comparación con los pacientes con hipertensión esencial de la misma edad, función renal y nivel de presión arterial [81]. Así, el tratamiento farmacológico antihipertensivo debería incluir un inhibidor del SRAA como primera opción [85,89].
La presión arterial en pacientes con PQRAD entre 18 y 50 años de edad que están en riesgo de progresión rápida (clasificación de la Mayo Clinic de 1C, 1D o 1E) debe apuntar a menos de 110/75 mmHg, ya que este control estricto de la presión arterial podría ralentizar la tasa de crecimiento de VRT y potencialmente enlentecer la disminución de TFGe. En todos los demás pacientes con PQRAD, la presión arterial puede tener un objetivo de <110/75 mmHg en jóvenes y <130/80 mmHg en mayores de 50 años [12,22,59].
Tolvaptán (antagonista del receptor V2)
El tolvaptán es un antagonista del receptor V2 (V2R) de la arginina vasopresina (AVP), con evidencia como terapia primaria para retrasar la progresión de la PQRAD. Se propone que el mecanismo de estos efectos implica la inhibición del V2R y la subsiguiente disminución de las concentraciones de 3',5'-monofosfato cíclico de adenosina en el riñón. Se cree que el 3',5'-monofosfato cíclico de adenosina elevado en el riñón promueve la acumulación de líquido en los quistes y el crecimiento de las células epiteliales, desplazando así al riñón normal y acelerando la aparición de ERC E5 [90].
El estudio de seguridad y eficacia de fase 3 de tolvaptán en PQRAD (TEMPO 3:4) demostró que el antagonismo de V2R con tolvaptán retrasó la tasa de crecimiento de VRT en un 49,2 % (de 5,5 % a 2,8 % por año). Es importante destacar que la TFGe se redujo en un 26 % (de 3,70 a 2,72 ml/min/1,73 m2 por año) durante los 3 años de seguimiento [91-94]. Un resultado secundario mostró que había un 36 % menos de riesgo de dolor renal informado por el paciente.
El estudio TEMPO 4:4 es un ensayo abierto de extensión en el que participaron 871 pacientes que habían completado el estudio TEMPO 3:4 (60,3 % de la muestra) en 106 centros de 13 países, donde se evaluó la eficacia y la seguridad del tratamiento durante dos años adicionales, de manera que los pacientes inicialmente tratados con tolvaptán continuaron el mismo tratamiento hasta por cinco años (grupo "tratamiento temprano") y los tratados con placebo cambiaron a tolvaptán durante dos años (grupo "tratamiento tardío"), la variable primaria fue el porcentaje de cambio desde el momento basal del estudio TEMPO 3:4 hasta el final del estudio TEMPO 4:4. No se observaron diferencias estadísticamente significativas en el porcentaje de VRT entre el grupo de "tratamiento temprano" (29,9 %) y el grupo de "tratamiento tardío" (31,6 %) (p = 0,38). Se observaron diferencias entre ambos grupos en la variable secundaria de cambio en la tasa estimada de filtrado glomerular (diferencia de 3,15 ml/min/1,73m2) (p <0,001), los resultados soportan el efecto de modificación de la enfermedad explicado por el tolvaptán en la TFGe [94,95].
En el ensayo REPRISE (Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptán Safety and Efficacy in ADPKD), los resultados confirmaron que el tolvaptán ralentizó la progresión de la enfermedad en PQRAD [12,93,96]. En este estudio multicéntrico, que incluyó 21 países, aleatorizado, doble ciego y comparado con placebo, se evalúo la eficacia y seguridad del tolvaptán en la reducción de la caída de la TFGe, donde se incluyó a un total de 1733 pacientes con edades entre 18 y 55 años, con una TFGe de 25 a 65 ml/min/1,73m2 y pacientes con edades de 55 a 65 años con TFGe de 25 a 44 ml/min/1,73m2, y encontró que al año de tratamiento con tolvaptán se presentó una reducción de la caída del TFG del 35 % (2,34 vs. 3,61 ml/min/1,73 m2 en el grupo tratado con tolvaptán vs. placebo) [96].
El estudio REPRISE incluyó a personas en etapa avanzada de PQRAD hasta la edad de 65 años y mostró una reducción similar y significativa en la disminución de la TFG-e [76, 96]; sin embargo, un análisis de subgrupos sugirió que este no era el caso para los pacientes mayores de 55 años, lo que implica que el tolvaptán solo debe ofrecerse hasta esta edad [76]. Adicionalmente, en una reciente publicación de Chebib et al. [97] se realizó un análisis agrupado con 230 participantes incluidos en 8 estudios, en el que se evaluaron los efectos del tolvaptán sobre la TFGe en pacientes con edades entre 56 y 65 años con ERC G3 o G4 y un declive de la TFG > 3 ml/min/1,73m2/año, en comparación con el tratamiento estándar; el estudio concluyó que se presentó una eficacia similar a la observada en la indicación general en los pacientes con menor edad [97], por lo que se requiere de nuevos estudios para dar recomendaciones en este rango de edad.
Con base en el ensayo REPRISE, se ha sugerido que el umbral inferior de TFGe para el inicio del tratamiento se reduzca a 25 ml/min/1,73 m2, ya que los análisis de subgrupos también muestran eficacia en esta etapa tardía [76].
La figura 6 sugiere un algoritmo de las intervenciones generales y farmacológicas en PQ- RAD.
Fuente: elaboración propia.
Figura 6 Algoritmo de las intervenciones generales y farmacológicas en PQRAD

Pregunta 9. ¿Cuáles son los criterios para iniciar tratamiento específico para la PQRAD con tolvaptán?
a clasificación de imágenes de la Mayo Clinic es una herramienta simple que utiliza el VRT ajustado a la altura y la edad, para identificar a los pacientes con mayor riesgo de progresión independientemente de la función renal [59].
En la mayoría de los pacientes, la ecuación elipsoide proporciona una estimación bastante precisa del VRT, la clase de imagen y la elegibilidad para el tratamiento [59].
La indicación aprobada por la FDA de tolvaptán es ralentizar el deterioro de la función renal en adultos con PQRAD rápidamente progresiva [12,59,98].
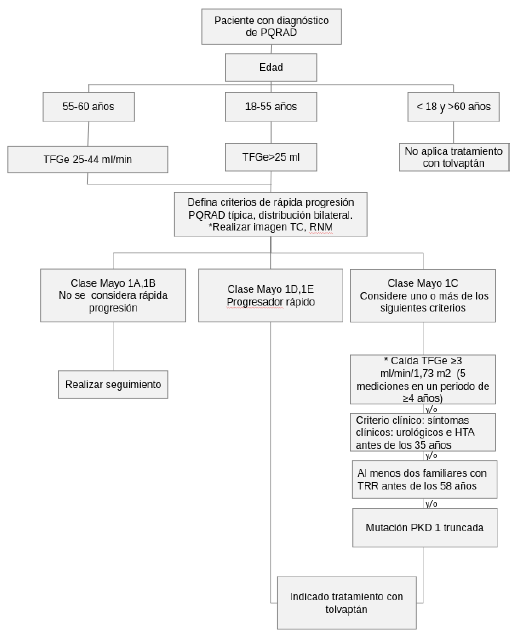
De acuerdo con lo descrito, se indica el tolvaptán en pacientes que tienen una tasa de filtración glomerular estimada (TFGe) ≥ 25 ml/min/1,73 m2 y se evidencian criterios de rápida progresión que han sido discutidos en la pregunta 6 [85,99].
Con estas consideraciones ajustadas al entorno y la evidencia actual, se propone el inicio del tratamiento con tolvaptán en pacientes con las siguientes características:
Pacientes con edad de 18-55 años, ERC estadio de 1 a 4 con TFGe ≥ 25 ml/min/1,73 m2 y evidencia de rápida progresión de clase 1D y E (clasificación Mayo Clinic).
Pacientes con edad de 18-55 años, ERC estadio de 1 a 4 con TFGe ≥ 25 ml/min/1,73 m2 y clase 1C (clasificación Mayo Clinic), que requiere evidencia adicional de rápida progresión, entre los cuales recomendamos utilizar uno o más de los siguientes criterios:
Si al menos dos familiares alcanzaron una terapia de remplazo renal antes de los 58 años.
Genéticos: mutación PKD1 truncada.
Inicio de temprano de síntomas clínicos (signos urológicos e hipertensión) antes de los 35 años de edad.
Disminución confirmada, fiable y basarse en al menos cinco mediciones del declive de la TFG-e > 3,0 ml/min por 1,73 m2 en un año durante cuatro años.
Paciente con PQRAD con edades de 56 a 60 años, con tasa de filtrado 25-44 ml/min/1,73 m2 con evidencia de rápida progresión de clase 1D, 1E y 1C (clasificación Mayo Clinic) (la clase 1C con evidencia adicional de acuerdo con la recomendación descrita).

Dosificación
El tolvaptán debe administrarse dos veces al día. La primera dosis suministrada en la mañana debe ser al menos 30 minutos antes del desayuno y la segunda dosis 8 horas después.
Se recomienda iniciar con una dosis de 60 mg/día hasta alcanzar los 120 mg/día [93].
Titulación
Una dosis inicial de 45 mg en la mañana y 15 mg ocho horas después y antes de las 4 pm para disminuir el riesgo de nicturia severa [59].
Se debe aumentar la dosis cada 1-4 semanas, según tolerancia, a un máximo de 90 mg en la mañana y 30 mg por la tarde [59] (tabla 10).
Tabla 10 Titulación de dosis de tolvaptán según tolerancia
| Dosis por semanas | 30 min antes del desayuno | 8 horas después (antes de las 4 pm) |
|---|---|---|
| Semana 1-4 | 45 mg | 15 mg |
| Semana 5-8 | 60 mg | 30 mg |
| Semana 9 en adelante | 90 mg | 30 mg |
Fuente: elaboración propia.

Pregunta 11. ¿En qué condiciones no se recomienda iniciar o continuar la terapia con tolvaptán?
Al tener en cuenta que el tolvaptán no está exento de efectos secundarios y que solo un grupo de pacientes cursará con una rápida progresión de la PQRAD y alcanzará una enfermedad renal avanzada, es necesario seleccionar adecuadamente qué pacientes pueden beneficiarse del tratamiento con tolvaptán y en qué casos debe suspenderse después de ser iniciado con base en sus contraindicaciones, efectos adversos y estilos de vida [47,59,100,101] (tabla 11).
Los principales efectos adversos relacionados con el tolvaptán se derivan de la acuaresis, la cual se expresa clínicamente como sed, polidipsia, poliuria, nicturia y polaquiuria (65-95 % de los casos), esta fue la principal razón para suspender el fármaco en el ensayo TEMPO 3:4, y la sed fue el evento adverso más común experimentado por más de la mitad de los pacientes [94, 96,102], sin embargo, estos síntomas se redujeron después de una exposición prolongada a la droga [103,104]. Con base en ello, debemos asegurarnos de que los pacientes que reciben este fármaco cuenten con un estado cognitivo que permita tener la capacidad para percibir o responder a la sensación de sed, un adecuado acceso al agua y la posibilidad de una excreción de grandes volúmenes de orina.
Un aspecto de gran importancia que debe considerarse con el uso del tolvaptán, es su potencial para inducir hepatotoxicidad idiosincrática, con elevación reversible de aminos-transferasas y potencial daño hepático grave. La prevalencia de hepatotoxicidad disminuye progresivamente con la exposición a tolvaptán [105], tal como se evidenció en los estudios TEMPO, donde la hepatotoxicidad se desarrolló dentro de los 3-18 meses de iniciado el tolvaptán.
En el estudio TEMPO 3:4, la incidencia de daño hepático definido como elevación de la alanina aminotransferasa (ALT) de tres veces el límite superior de la normalidad (LSN), fue del 4,4 % en los pacientes que recibieron tolvaptán vs 1,0 % de los tratados con placebo y una elevación >3 veces el LSN del aspartato aminotranferasa (AST) en el 3,1 % de los tratados con tolvaptán vs. 0,8 % del grupo placebo, siendo más frecuente estas elevaciones en los primeros 18 meses. La incidencia de eventos hepáticos fue similar en el estudio TEMPO 4:4 en el grupo de tratamiento tardío respecto al grupo de tratamiento temprano; sin embargo, hay reportes de casos que sugieren que la hepatotoxicidad idiosincrásica asociada al tratamiento con tolvaptán es más común en la práctica médica que lo reportado en la literatura [106], por lo que se debe ser estricto al monitoreo de las pruebas hepáticas.
La monitorización de marcadores de daño (transaminasas) y función hepática (bilirrubinas), permite prever el desarrollo de esta complicación, siendo necesaria realizar la medición de estos marcadores cada mes durante los primeros 18 meses de tratamiento y cada 3 meses a partir de los 18 meses [4].
Un aspecto para tener presente son las posibles interacciones farmacológicas, ya que el tolvaptán no debe ser usado de forma concomitante con inhibidores potentes de la citocromo CYP3A y su dosis debe ser reducida cuando se utilizan inhibidores moderados de este complejo enzimático. De igual forma, el tolvaptán podría elevar los niveles de los sustratos transportadores OATP1B1/3 y OAT3 y los sustratos transportadores de BCRP, por lo que su uso concomitante con dichos fármacos generalmente debería ser evitado (tabla 12) [59].
Además, se recomienda evitar el consumo de zumo de pomelo, ya que aumenta las concentraciones plasmáticas del medicamento.
Con relación a las estatinas, aproximadamente el 14 % de los pacientes asignados al azar a tolvaptán en el ensayo TEMPO 3:4 fueron tratados con estos fármacos y no se detectó asociación con toxicidad hepática, sin embargo, se deben usar las estatinas con precaución y solo cuando esté claramente indicado.
En la actualidad no hay datos suficientes para determinar el riesgo de tolvaptán para el desarrollo fetal, por lo que está contraindicado en el embarazo y la lactancia.
Tabla 11 Cuando no iniciar tolvaptán
| Criterios | Condiciones |
|---|---|
| Pacientes |
|
| Volumen |
|
| Bioquímicas | • ALT o AST >3 veces y BT >2 veces LSN o INR >1,5 o signos de daño hepático |
Nota: ALT: alanina transaminase; AST: aspartato transaminasa; BT: bilirrubina total; CP: concentración plasmática; INR: international normalized ratio; LSN: límite superior normal; TFGe: tasa de filtración glomerular estimada.
Fuente: elaboración propia.
Una vez iniciada la terapia con tolvaptán, las pruebas de laboratorio para evaluar su seguridad incluyen: AST, ALT, fosfatasa alcalina, bilirrubina total, sodio, creatinina y ácido úrico. De igual forma, es necesario educar a los pacientes sobre no automedicarse o ingerir preparados herbales, y estar atentos de la aparición de cualquier síntoma o signo de daño hepático: fatiga, náuseas, vómitos, dolor o sensibilidad en el cuadrante superior derecho, ictericia, fiebre y erupción cutánea, lo que obligaría a la suspensión inmediata del fármaco y a repetir pruebas en 48-72 horas.
Tabla 12 Interacciones medicamentosas relevantes
| Interacciones medicamentosas | |
|---|---|
| Aumentan CP: inhibidores del CYP3A4 | Rifampicina, barbituricos, rifabutina, rifapentina, fenitoina, carbamazepina, hiperico o hierba de San Juan |
| Aumentan CP: inhibidores del CYP3A4 | Potentes: ketoconazol, itraconazol, lopinavir, ritonavir, indinavir y claritromicina. Moderados: amprenavir, aprepitant, atazanavir, ciprofloxacina, crizotinib, darunavir/ritonavir, amiodarona, eritromicina, fluconazol, diltiazem, verapamilo, imatinib, fosamprenavir |
| Elevación de niveles transportadores OATP1B1/3 y OAT3 | Estatinas, furosemida, gliburida, repaglinida, metotrexato |
| Elevación sustrato transportadores BCRP | Rosuvastatina |
Nota: CP: concentración plasmática.
Fuente: elaboración propia.
Si se resuelven las alteraciones de laboratorio, se puede reiniciar el tolvaptán con una mayor frecuencia de control (semanalmente durante el primer mes), siempre que la ALT y la AST se hayan mantenido por debajo de tres veces el LSN (límite superior normal); sin embargo, el tolvaptán debe suspenderse de forma permanente en pacientes sin evidencia de otras causas de daño hepático (hepatitis aguda o agentes hepatotóxicos concomitantes), excepto tolvaptán y con resultados de laboratorio de seguimiento: AST o ALT >3 veces el LSN y la bilirrubina >2 veces el LSN, AST o ALT >5 veces el LSN durante dos semanas o AST o ALT >8 veces el LSN [107].
En relación con la natremia, esta debe ser evaluada de manera simultánea con las pruebas de función hepática y es fundamental su seguimiento para así determinar requerimientos de ingesta de líquidos y ajuste de dosis de tolvaptán (tabla 13).
Adicionalmente, el uso de tolvaptán se ha relacionado con un aumento de los niveles séricos de ácido úrico, donde los pacientes que recibieron tolvaptán experimentaron con más frecuencia hiperuricemia (3,9 % frente a 1,9 %) y gota (2,9 % frente a 1,4 %) en comparación con el grupo de placebo, con una aumento progresivo en los niveles de ácido úrico según avanza la enfermedad renal [102].
Es probable que el uso concomitante de diuréticos y tolvaptán aumente el riesgo de gota, por lo que se debe evitar el uso concomitante de estos medicamentos [108]. Además, deben controlarse los niveles de ácido úrico sérico y se debe considerar un agente reductor del ácido úrico para disminuir el riesgo de gota si el nivel de ácido úrico supera los 10 mg/dl o para tratar la afección si se desarrolla. En la actualidad, no hay pruebas suficientes para recomendar el tratamiento de la hiperuricemia asintomática [59].
Tabla 13 Indicaciones para suspender o ajustar la dosis de tolvaptán
Nota: ALT: alanina transaminasa; AST: aspartato transaminasa; BT: bilirrubina total; INR= international normalized ratio; LSN: límite superior normal; TFGe: tasa de filtración glomerular estimada; TRR: terapia de reemplazo renal; Na: sodio.
Fuente: elaboración propia.

12a) Para evaluar progresión de la enfermedad
La eficacia del tratamiento generalmente se controla mediante la graduación de la disminución de TFGe, la tasa de crecimiento de VRT y la calidad de vida.
No se recomienda la medición rutinaria del VRT como parámetro para evaluar la eficacia del tolvaptán en el manejo de la PQRAD, sin embargo, puede ser útil obtener una RMN o TC para medir el VRT cada 3-5 años para evaluar la tasa de crecimiento de VRT al compararla con la de imagen inicial obtenida del paciente [75]. En caso de deterioro súbito de la función renal, puede considerarse la realización de una RMN para descartar otras causas que expliquen el descenso del filtrado glomerular.
Por otra parte, se recomienda la medición de la creatinina sérica para calcular la TFGe como determinante de la efectividad del tratamiento. Es importante determinar la TFGe basal al inicio del tratamiento como referente y compararla con los cambios anuales en la misma, para establecer así la tendencia del compromiso de la función renal (declinación vs. estable).

12b) Prevención y manejo de efectos adversos al tolvaptán (control de función hepática) Los principales efectos secundarios del tratamiento con tolvaptán incluyeron hepatotoxicidad, poliuria, polaquiuria, nicturia, sed y fatiga [103,109].
El sodio plasmático debe mantenerse de manera óptima entre 135 y 143 mmol/l. El sodio aumenta en promedio 2,5 mEq/l, alcanzando niveles significativos (>150 mEq/l) en 4 % de los pacientes tratados con tolvaptán. Por otra parte, los pacientes deben tener acceso al agua e ingerirla aun cuando no sientan sed, además, se debe instruir al paciente en prevenir la deshidratación e ingerir en promedio cuatro litros al día para evitar la hipovolemia.
Se ha sugerido evaluar la osmolalidad urinaria en la primera orina de la mañana antes de la primera toma del tolvaptán para confirmar la ingesta de este y la obtención de su objetivo en los pacientes al inicio y en el seguimiento del tratamiento. Una reducción por debajo de 275- 280 mOsm/kg se considera ideal en la mayoría de los casos [47], donde el logro de esta osmolalidad asegura la dosis mínima para inhibir la vasopresina [59].
Los efectos menos frecuentes pero los más severos son los efectos hepáticos con aumento de las transaminasas, que implican la suspensión de la medicamento en el 2,1-3,6% de los pacientes [43, 77] con hepatotoxicidad idiosincrática, esto ocurre generalmente durante los primeros 18 meses de tratamiento, generalmente es reversible y no debe suspenderse el tratamiento tempranamente, salvo en las recomendaciones que se han determinado para suspender (tabla 13) (pregunta 11, tabla 13). La injuria hepática e irreversible ha sido extremadamente rara y, teniendo en cuenta la posible toxicidad hepatocelular, se debe monitorear la función hepática antes de iniciar el tratamiento con tolvaptán y después a las dos y cuatro semanas, luego validar mensualmente durante 18 meses y luego cada3 meses [107,110].

Pregunta 13. ¿Qué otros tratamientos farmacológicos han demostrado eficacia para evitar la progresión de la PQRAD?
Los avances significativos en la comprensión de la genética de la PQRAD y los mecanismos de crecimiento de los quistes han revelado objetivos probables adicionales para la intervención terapéutica [111]. Dentro de estas dianas terapéuticas fueron investigados:
Los análogos de la somatostatina que de manera individual no demostraron impacto en la progresión de la PQRAD, en el estudio DIPAK1, aunque la tasa de crecimiento del VRT fue más baja y no mostró diferencias significativas en la disminución anual de la TFGe, además, resultados similares fueron encontrados en el estudio ALADIN 2 [13,112, 113]; sin embargo, los análogos de la somatostatina de acción prolongada (octreotida de acción prolongada (LAR)) asociados a tolvaptán tienen efectos renoprotectores en la PQRAD, que están parcialmente mediados por la mejora de la hiperfiltración glomerular compensatoria [114,115].
Los agentes antiproliferativos inhibidores de los mTOR e inhibidores de la tirosin- quinasa no mostraron cambios en el VRT ni en la TFGe [13,116,117].
La metformina afecta la cistogénesis en modelos preclínicos de enfermedad poliquística, pudiendo ser una terapia prometedora para la investigación clínica en PQRAD. En el estudio TAME- PKD aleatorizado, doble ciego, no se encontraron diferencias significas en la disminución TFGe [118,119]. El uso concomitante de metformina hidroclorotiazida y tolvaptán ha mostrado la reducción de la poliuria causada por tolvaptán sin afectar la nefroprotección, pero estos resultados requieren estudios complementarios, dado que el ensayo clínico solo incluyó 13 pacientes [120].
Activador del factor 2 relacionado con el factor eritroide 2 (Nrf2), el cual se encuentra en fase III de investigación con bardoxolona metil (BARD), un activador de Nrf2. La activación de Nrf2 disminuye el estrés oxidativo y se ha demostrado que es valiosa en
la ERC [13].
El venglustat, que es un inhibidor de la glucosilceramida sintasa, en ensayos preclínicos logró inhibir el crecimiento de quistes renales y enlentecer la progresión del daño renal, sin embargo, en el estudio STAGED-PKD que fue realizado con el objetivo de determinar la seguridad y la eficacia de venglustat en pacientes con PQRAD con criterios de rápida progresión, el tratamiento a dosis de 8 mg o 15 mg no mostró cambios en la tasa de crecimiento del VRT y se evidenció una rápida disminución de la TFGe, por lo que el estudio se terminó antes de tiempo [121, 122].

Pregunta 14. ¿Qué alternativas de tratamiento sustitutivo renal están indicadas en el paciente con PQRAD y qué evidencia respalda su utilización en la actualidad?
La PQRAD es la enfermedad genética que más conduce a enfermedad renal en Europa y el resto del mundo [123,124]. Además, representa el 5-10 % de todos los pacientes que requieren terapia renal sustitutiva, entre ellas diálisis peritoneal, hemodiálisis o trasplante renal [47,125].
Los pacientes con PQRAD en terapia sustitutiva tienen una supervivencia más alta que la de los pacientes no afectados de PQRAD.
La diálisis peritoneal (DP) está infrautilizada en la PQRAD, porque históricamente se ha pensado en fallas técnicas causadas por la reducción del espacio de la cavidad abdominal y el incremento de la presión intraabdominal [126]. Así lo muestra el Registro Francés REIN (Renal Epidemiology and Information Network), donde se comparan las características de los pacientes tratados con diálisis peritoneal vs. hemodiálisis, solo el 10,9 % de la población con PQRAD fue tratada con DP [127]. Estudios recientes han demostrado una supervivencia global similar a los pacientes tratados con DP y los que reciben hemodiálisis, y con tasa de supervivencia similar a los pacientes sin PQRAD. Además, parece ser similar con respecto a la técnica y las tasas de peritonitis en pacientes con PQRAD en comparación con individuos con ERC sin PQRAD [79]. Las guías españolas han sugerido que la diálisis peritoneal oferta un mejor pronóstico y calidad de vida a los pacientes con PQRAD que a aquellos que no son portadores de esta enfermedad, sin embargo, en pacientes con riñones o hígados muy grandes, la ausencia de espacio puede limitar el área disponible para el intercambio peritoneal y aumentar las posibilidades de hernias abdominales e hidrotórax, en estos casos debería considerarse la hemodiálisis como mejor opción, al igual que para aquellos pacientes con diverticulitis recurrente [47].
La elección de la modalidad de diálisis debe ser individualizada, de acuerdo con los antecedentes y el deseo del paciente, al soporte social y a la comorbilidad, como se hace en las personas con otras causas de enfermedad renal [47].
El trasplante renal en pacientes con PQRAD tiene tasa de efectividad y complicaciones similares a la que presentan los pacientes trasplantados con ERC por otras causas [123,128]. Otros grupos (Clinical Practice Guideline for Autosomal Dominant Polycystic Kidney Disease in China) recomiendan que el trasplante renal sea la opción óptima de terapia de reemplazo renal para pacientes con ERC E5 por PQRAD [22]. Además, el trasplante renal de donante vivo de forma preventiva se ha asociado con una mejor evolución.

Pregunta 15. ¿Cómo se debe preparar un paciente con PQRAD candidato a trasplante renal?
El trasplante renal es la mejor opción de tratamiento renal sustitutivo, la tasa de efectividad y las complicaciones de pacientes trasplantados con PQRAD y falla renal es la misma que en los pacientes que la presentan por otras causas y no es factor de riesgo para el desarrollo de diabetes postrasplante [15].
Durante el estudio pretrasplante del paciente con PQRAD, se sugiere considerar los siguientes puntos [15]:
Nefrectomía rutinaria antes del trasplante. En la actualidad, solo en menos de 1/3 de los pacientes es realizada (ver pregunta 16) y ya no se recomienda de forma rutinaria [129,130].
Embolización arterial transcatéter de arterias renales. Los riñones severamente agrandados son una contraindicación para trasplante por falta de espacio disponible en la pelvis y mayores tasas de fracaso del trasplante. La nefrectomía pretrasplante proporcionará espacio dentro de la pelvis y sirve para disminuir los síntomas de compresión asociados con un riñón agrandado, sin embargo, la nefrectomía es un procedimiento quirúrgico invasivo con una gran variedad de complicaciones que incluyen hemorragias graves, hernia, desarrollo de fístulas arteriovenosas, trombosis, lesión vascular y riesgo de infección. Además, puede comprometer el acceso de diálisis peritoneal y dados estos riesgos hay una alternativa no invasiva para reducir el volumen renal: la embolización arterial transcatéter de las arterias renales, el cual es un procedimiento no invasivo y bien tolerado con efectos secundarios mínimos [131].
Tamizaje para detección de aneurismas intracerebrales. Las guías australianas "CARI" de aneurismas intracerebrales recomiendan hacer tamizaje de aneurisma intracerebral en pacientes con PQRAD ante los siguientes criterios [79]
Historia familiar en al menos un pariente de primer grado que haya tenido hemorragia subaracnoidea, hemorragia intracerebral o aneurisma.
Pacientes con cefalea intensa y súbita o cambios en el estado neurológico. Se realiza el tamizaje con angiografía del círculo de Willis, ya sea por RMN o TC, cada 5 o 10 años después del primer estudio con resultado negativo [15].
Tamizaje para detección de quistes intrahepáticos con TC o RMN y la medición de CA 19-9. Se debe determinar si los pacientes con PQRAD que son candidatos para trasplante renal reportan quistes intrahepáticos, con el fin de evaluar la posibilidad de llevar a cabo un trasplante dual renohepático, el cual es una opción en casos de colangitis recurrente o hepatomegalia dolorosa, cuando el paciente presenta TFG ajustada por altura mayor o igual a 30 ml/minuto/1,73 m2.
Inicio de hemodiálisis inmediatamente después de nefrectomía, si esta se practica previa a trasplante.

Pregunta 16. ¿Cuándo está indicada la nefrectomía en pacientes con PQRAD?
La nefrectomía pretrasplante está indicada cuando se requiere suficiente espacio para el implante del injerto, que generalmente se anastomosa en la fosa ilíaca derecha. Debido al gran desorden anatómico inducido por el volumen, la identificación del uréter es la máxima prioridad para encontrar el plano adecuado para la nefrectomía [131,132]. No hay suficiente evidencia acerca de si la nefrectomía del riñón nativo debe realizarse pretrasplante o simultánea en el acto del trasplante, y esta dependerá de si el riñón es sintomático. La nefrectomía laparoscópica es una buena opción, siempre que el centro tenga experiencia en este tipo de cirugía.
Otras indicaciones de nefrectomía antes del trasplante incluyen: dolor severo, saciedad temprana, infección severa y recurrente en los quistes renales, hemorragias recurrentes o graves, dolor intenso, nefrolitiasis sintomática y sospecha de cáncer renal [129,131].

Pregunta 17. ¿Cuáles son los criterios para ser candidato a donante vivo relacionado para un paciente con PQRAD?
El trasplante renal en la PQRAD es la mejor opción de terapia de reemplazo sustitutivo y con donante vivo de forma preventiva muestra una mejor evolución.
Los familiares del paciente con PQRAD que quieran donar su riñón deben cumplir los siguientes criterios establecidos desde el estudio pretrasplante de donantes:
Si tienen menos de 30 años deben contar con estudios genéticos que reporten un resultado negativo de la mutación PKD1 o PKD2 e imagen de RMN sin alteraciones.
Donantes con edades entre 30 y 40 años deben tener imagen de RMN negativa para enfermedad quística.
Los donantes mayores de 40 años deben de tener uno o ningún quiste renal [15].

Recomendación de estudios genéticos
El estudio genético no está indicado a todos los pacientes, pero existen consideraciones para las familias en los siguientes casos:
Determinar patrón de herencia y determinar el riesgo de PQRAD en sus familiares.
Consejo genético para las familias con antecedentes familiares que desean tener hijos, con fines de posibilidad de pruebas de preimplantación genética.
Pacientes sin antecedentes familiares de PQRAD y con hallazgos imagenológicos con características atípicas que ameriten descartar otras enfermedades quísticas.
La PQRAD es una enfermedad autosómica dominante, con un 50 % de posibilidad de que los hijos sean afectados, por lo que es natural que muchos de los familiares expresen su preocupación. El estudio genético no está indicado en todos los pacientes, pero entre los familiares que deseen consejo genético con fines de conocer el riesgo previo a tener hijos o aquellos que lo deseen para fines de preimplantación, la realización de dichas pruebas es razonable. Menos del 20 % consideraran terminación del embarazo basado en los estudios genéticos, pero el 70 % desean pruebas genéticas preimplantación para bloquear la transmisión de mutaciones de PQRAD patogénicas y disminuir la posibilidad de nacimiento de hijos afectados [22,133].
¿Cuándo realizarse estudios de imágenes diagnósticas en los familiares de afectados?
La ultrasonografía es el estudio de tamizaje por excelencia en los familiares de pacientes con PQRAD para llegar a un diagnóstico temprano [134].


