










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.53 no.4 Bogotá Oct. 2005
Actualización
Carlos Olimpo Mendivil Anaya1, Iván Darío Sierra Ariza2
1.Médico con entrenamiento en Lípidos y Diabetes, Especialista en Estadística. Profesor Adjunto e Investigador, División de Lípidos y Diabetes, Facultad de Medicina, Universidad Nacional de Colombia Bogotá.
2Médico especialista en Diabetes y Nutrición, Profesor Titular y Maestro Universitario, Coordinador División de Lípidos y Diabetes, Facultad de Medicina, Universidad Nacional de Colombia Bogotá
Correspondencia: comendivila@unal.edu.co
Resumen
Las patologías asociadas a alteraciones metabólicas crónicas, como la obesidad, la diabetes y las dislipidemias; constituyen una fuente de enfermedad, muerte y discapacidad que cobra cada vez mayor relevancia en el mundo. Varios de estos problemas son consecuencia directa o indirecta de una acción insuficiente de la insulina, lo cual origina una constelación de factores de riesgo que potencian la aparición de enfermedad cardiovascular aterosclerótica en el largo plazo.
En esta revisión se abordan de manera concisa los mecanismos de acción de la insulina a nivel molecular, así como la forma en que se pueden ver afectados por factores diferentes a la constitución genética de cada individuo.
Palabras clave: insulina, diabetes mellitus, metabolismo, fosforilación.
Summary
Pathologies associated with chronic metabolic disturbances like obesity, diabetes and dyslipidemia, constitute major contributors to the worldwide burden of disease, death and disability, and are acquiring a higher relevance every day. Several of these problems are a direct or indirect consequence of an insufficient action of insulin, which gives rise to a cluster of risk factors that potentiate the onset of cardiovascular atherosclerotic disease in the long term.
This review adresses in a concise way the mechanisms of insulin action at the molecular level, as well as the way they can be affected by factors different to the genetic constitution of each individual.
Key word: insulin, diabetes mellitus, metabolism, phosphorylation.
Introducción
El incremento agudo en la incidencia de enfermedades crónicas no transmisibles, especialmente obesidad, diabetes y enfermedades cardiovasculares ateroscleróticas, ha generado en la comunidad científica médica un interés sin precedentes por descifrar sus bases fisiopatológicas.
En ese sentido, la comprensión de la regulación metabólica reviste una gran importancia; incluyendo el entendimiento de los mecanismos moleculares de acción hormonal. En este artículo se busca resumir de manera global los mecanismos moleculares de acción insulínica, y la forma como se pueden ver afectados por condiciones diferentes a la constitución genética del individuo.
Acción insulínica: la insulina posee efectos en múltiples órganos blanco que se podrían clasificar en agudos; que afectan principalmente el metabolismo de carbohidratos y en efectos intermedios y a largo plazo.
Los principales efectos agudos de la insulina sobre los tejidos blanco son (mas adelante se detalla cómo induce la insulina tales efectos) (1):
Estímulo de la captación de glucosa, mediante el favorecimiento de la traslocación de los glucotransportadores GLUT-4 a la membrana plasmática en músculo y tejido adiposo (1,2).
Estímulo de la síntesis de glucógeno e inhibición de su degradación en hígado y músculo (1,2).
Estímulo del metabolismo oxidativo de la glucosa (glucólisis) (1,2).
Inhibición de la gluconeogénesis hepática (1,2).
Estímulo de la captación y almacenamiento de grasas por el tejido adiposo (estímulo a la LPL-1 y triglicérido sintasa) (1,2).
Inhibición de la lipólisis en tejido adiposo (por inhibición de la lipasa adipolítica u hormonosensible) (1,2).
Los principales efectos a mediano y largo plazo de la insulina son:
Efectos sobre la captación/retención de iones y el metabolismo hidroelectrolítico (3).
Estímulo a la síntesis e inhibición de la degradación de proteínas (3).
Efectos sobre la expresión génica (trascripción) (3).
Efectos sobre el recambio del mRNA (3).
Estímulo del crecimiento, proliferación y diferenciación celulares (3).
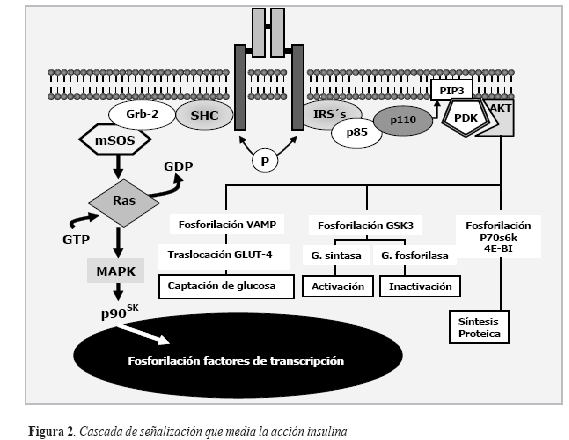
Bioquímica de la acción insulínica: las moléculas implicadas en el proceso de la respuesta biológica a la insulina se pueden clasificar en tres niveles (4):
Nivel 1: desde el receptor de insulina, hasta el nivel correspondiente a la fosfatidilinositol 3-cinasa (PI3K).
Nivel 2: moléculas todas que actúan como segundos mensajeros intracelulares, la mayoría involucradas en fenómenos de fosforilación secuencial.
Nivel 3: moléculas involucradas en la respuesta efectora a la insulina.
Examinemos la secuencia que sigue a la unión de la insulina a su receptor, comenzando con las moléculas del Nivel 1.
Receptor de Insulina
El receptor de insulina es una proteína tetramérica con dos subunidades alfa extracelulares y dos subunidades beta que tienen una pequeña porción extracelular, una porción transmembranal y una porción intracelular (o intracitoplásmica) (5). Los tejidos con mayor abundancia de receptores de insulina son el parénquima hepático y el tejido adiposo, donde pueden llegar a existir 200,000 a 300,000 copias del receptor por célula.
Las dos subunidades del receptor proceden de un mismo gen y un mismo trascripto, que en el aparato de Golgi sufre glucosilación, acilación y posterior proteólisis para generar el receptor definitivo con sus cuatro subunidades.
El receptor de insulina puede entenderse como una enzima alostérica en la cual las subunidades beta son las subunidades catalíticas y las subunidades alfa son subunidades regulatorias que las mantienen inhibidas. Cuando la insulina se une a las subunidades alfa, la actividad inhibitoria de éstas sobre las subunidades beta se pierde (Figura 1) (5,6). En ese momento las subunidades beta ejercen su acción catalítica de tirosín-cinasas, las dos subunidades se transfosforilan (la una fosforila a la otra y viceversa) en 6-7 residuos de tirosina. Sin ésta actividad tirosín-cinasa del receptor de insulina, no se da ninguno de los efectos biológicos de la insulina.

Las subunidades beta también poseen residuos de serina y treonina que se pueden fosforilar (5,6). Cuando esto sucede, la actividad tirosín-cinasa se reduce notablemente y todos los efectos insulínicos se disminuyen. La fosforilación en serina y treonina de las subunidades beta del receptor de insulina ejerce una acción regulatoria negativa sobre la respuesta biológica a la insulina (5,6,7).
Sustratos del receptor de insulina
En algún tiempo se pensó que la acción insulínica estaba mediada por cambios en la concentración intracelular de nucleótidos (AMPc, GMPc) o flujos iónicos, pero hoy se sabe que la mayoría de sus efectos están mediados por la fosforilación de sus sustratos endógenos (IRS, de Insulin Receptor Substrates). Se han identificado cuatro IRS, pero los más estudiados han sido el IRS-1 y el IRS-2, que son ubicuos, mientras que IRS- 3 está restringido de tejido adiposo, e IRS-4 a riñón y encéfalo (8).
El IRS-1 es una proteína rica en regiones de unión a tirosinas fosforiladas (regiones PTB, de PhosphoTyrosine Binding), que le permiten unirse al receptor y ser fosforilado por él en sus residuos de tirosina (8,9). Una vez el IRS-1 es fosforilado, liga a dos moléculas de gran importancia en la respuesta biológica a la insulina: PI3K y Grb-2 (1,8,9).
Si bien no se ha podido esclarecer del todo la importancia relativa y las funciones de IRS-1 e IRS-2, estudios realizados con knock-out genéticos de ambos genes han mostrado que en términos generales los animales sin IRS-1 tienen baja talla, peso y desarrollo; mientras que los animales sin IRS-2 son diabéticos e insulinorresistentes (10-12).
Así, al parecer IRS-1 media primordialmente los efectos "tróficos", de la insulina; mientras que IRS-2 está más involucrado con los efectos "metabólicos" de la hormona.
Fosfatidilinositol 3-cinasa (PI3K)
PI3K es probablemente la enzima de la cascada de señalización de insulina más extensamente estudiada. Es una proteína dimérica con una subunidad catalítica (p110) y una subunidad regulatoria (p85). La subunidad p85 se une a los IRS fosforilados y eso hace que cese su actividad inhibitoria sobre la subunidad p110 (1,13,14).
a subunidad p110 desinhibida fosforila varios fosfolípidos de membrana, principalmente el fosfatidilinositol 4,5 bifosfato (PI 4,5P) para generar fosfatidilinositol trifosfato (PIP3). El PIP3 es el encargado de fijar a la membrana y activar a PDK1 y AKT, dos enzimas cinasas que median la mayoría de los efectos metabólicos de la insulina (1,15).
Cuando se han realizado experimentos con bloqueo genético o farmacológico de la actividad de PI3K, sucede lo siguiente (16-19):
Se reduce dramáticamente la expresión de GLUT-4 en la membrana plasmática. Se pierde el "freno antilipólisis" brindado por la insulina. Se inactiva la glucógeno sintetasa. Se reduce de forma importante la síntesis de nuevas proteínas y DNA.
Grb-2
Los IRS fosforilados también ligan una proteína llamada Grb-2, que tras su unión con los IRS se dimeriza con la proteína mSOS (1,20). El complejo Grb-2/mSOS actúa sobre una proteína GTPasa asociada a la membrana llamada Ras, haciendo que intercambie GDP por GTP y se active toda la vía de las MAP cinasas, esencial en la regulación del crecimiento y proliferación celulares, así como de la expresión génica (21).
¿Cómo ocasiona la activación de PI3K los efectos metabólicos de la insulina?, debemos examinar las acciones de niveles 2 y 3.
PDK1/AKT
Después de que PDK1 y AKT se han fijado a la membrana y AKT se encuentra activo, AKT fosforila varias proteínas ocasionando efectos metabólicos importantes:
- AKT fosforila a VAMP y otras proteínas de fusión presentes en las vesículas de almacenamiento de los GLUT-4, ocasionando la traslocación de los GLUT-4 a la membrana y por tanto la captación de glucosa (22).
- AKT fosforila a GSK3, una enzima que fosforila a la glucógeno sintasa y a la glucógeno fosforilasa. Cuando AKT fosforila a GSK3, la inactiva. Así, habrá una menor fosforilación tanto de la glucógeno sintasa como de la glucógeno fosforilasa (1,23,24).
La glucógeno sintasa sin fosforilar es activa, mientras que la glucógeno fosforilasa sin fosforilar es inactiva. Se dará por tanto una mayor síntesis y menor degradación del glucógeno.
- AKT fosforila a varias proteínas ribosomales, entre ellas p70S6K y 4E-BI, un factor de inicio de la traducción, activándolo. Se estimulará por tanto la síntesis de proteínas (25).
La secuencia de señalización intracelular de la insulina se resume globalmente en la figura 2.
Influencia del medio ambiente sobre la acción de la insulina
Después de conocer la intrincada red que regula la respuesta celular a la insulina, y sabiendo que son factores en su mayoría medioambientales los que inducen resistencia a la insulina, surge la pregunta: ¿Cómo puede el medio ambiente inducir resistencia a la insulina?
Sobrepeso y obesidad
En pacientes con diabetes tipo 2 obesos, así como en obesos no diabéticos pero insulinorresistentes, se ha evidenciado una reducción sustancial en la actividad tirosín-cinasa del receptor de insulina (26-27).
¿Está la reducción en la actividad tirosín-cinasa del receptor determinada genéticamente? Al parecer no, dado que la actividad puede ser notablemente diferente entre gemelos monocigóticos (28).
¿Existen mutaciones puntuales "esporádicas" que reduzcan la actividad tirosín-cinasa del receptor y sean responsables de la diabetes? Aunque se han descrito mutaciones del receptor que afectan su actividad de tirosín-cinasa, su frecuencia en pacientes con diabetes tipo 2 es sumamente baja, así que es muy poco probable que jueguen un papel determinante en la génesis de la enfermedad en la mayoría de los pacientes (29).
¿La obesidad induce una reducción "irreversible" en la actividad tirosín-cinasa del receptor?
No, de hecho se ha demostrado que la actividad de tirosín-cinasa se recupera completamente en pacientes con diabetes tipo 2 obesos que llegan al normopeso (30).
Cuando el nivel de adiposidad corporal se incrementa, especialmente dentro de la cavidad abdominal, se genera un estado de disfunción adipocitaria caracterizado por adipocitos, grandes, resistentes a la insulina y productores de hormonas capaces de inducir resistencia a la insulina en los tejidos vecinos y a distancia en otros tejidos (31-35).
Los adipocitos disfuncionantes son resistentes a la acción antilipolítica de la insulina y están liberando constantemente ácidos grasos libres a la circulación sistémica (31-35).
Los ácidos grasos libres llegan a músculo e hígado e inducen la producción de di-acil glicerol (DAG), un segundo mensajero que activa a una familia de serina-treonina cinasas conocidas en conjunto como Proteín Cinasa C (PKC) (36). Recordemos que la fosforilación del receptor de insulina en serina y treonina disminuye su actividad de tirosín-cinasa y por ende toda la respuesta celular a la insulina, de ahí que cuando el receptor es fosforilado en serina y treonina por la PKC, se induce resistencia a la acción de la insulina. Así, es posible afirmar que parte de la resistencia a la insulina ocasionada por el sobrepeso es mediada por los ácidos grasos libres, como activadores indirectos de serina-treonina cinasas.
En cuanto a los sustratos del receptor de insulina (IRS´s), se ha encontrado que su fosforilación se encuentra inalterada en humanos con diabetes tipo 2 (descontando el efecto de la menor actividad de tirosín-cinasa del receptor) (36). Sin embargo sí se ha evidenciado una reducción de 3-4 veces en la activación de PI3K en pacientes con diabetes tipo 2 (37-38), un hallazgo congruente con el tipo de alteración fisiológica encontrada en ellos: una respuesta preservada a los efectos tróficos de la insulina, pero una respuesta fuertemente alterada a los efectos metabólicos de la misma.
Factor de necrosis tumoral alfa (TNF- )
El TNF-a, identificado inicialmente en macrófagos y llamado caquectina, es producido también por el tejido adiposo y cumple esencialmente una función paracrina (sobre células adyacentes) y autocrina (sobre el propio tejido adiposo). Existen dos tipos de receptores de TNF-a (TNFR-1 y TNFR-2) y el tejido adiposo expresa ambos (39).
En algunos estudios se ha encontrado correlación entre los niveles plasmáticos del TNF-a y la resistencia a la insulina, pero en otros no. Probablemente ésta aparente contradicción se deba a que sólo una pequeña fracción del TNF-a que es secretado sale a la circulación general, la mayor parte cumple su función en el propio tejido adiposo o en el músculo adyacente o cercano al tejido adiposo y es degradado in situ. En ese sentido es importante señalar que muchos de los efectos deletéreos de la obesidad sobre la salud se deben a que acerca los adipocitos al músculo (39,40).
Cuando el TNF-a se une a su receptor en hígado, se desencadena una cascada de señalización actualmente objeto de intenso estudio, que conduce a un estímulo en la síntesis de colesterol y ácidos grasos tanto en tejido adiposo como en hígado, activación de serina-treonina cinasas, especialmente la isoforma epsilon de la PKC (PKC-e) (40).
El bloqueo en la señalización intracelular de insulina inducido por el TNF-a se traduce en inhibición de las enzimas involucradas en la captación de ácidos grasos, en la captación de glucosa y en la síntesis de triglicéridos; causando por tanto hiperglucemia e incremento en la concentración de ácidos grasos libres en sangre (40).
Así pues, otro de los probables nexos entre sobrepeso y resistencia a la insulina está dado por el TNF-a, una citocina capaz de antagonizar efectivamente la acción insulínica.
Adiponectina
La adiponectina es una hormona producida específicamente por el tejido adiposo y, a diferencia del TNF-a, la mayor parte de su producción llega a la circulación sistémica (41). Se expresa más en tejido adiposo subcutáneo que en tejido adiposo visceral, y su concentración se incrementa cuando la sensibilidad a la insulina mejora. Para que tenga adecuada actividad biológica, la adiponectina debe estar hidroxilada y glucosilada, lo cual genera varias isoformas de acuerdo al grado de hidroxilación y glucosilación (42-43).
Se han identificado dos receptores diferentes de adiponectina: El receptor AdipoR1, que se expresa primordialmente en músculo, y el AdipoR2, que lo hace en hígado (43). El AdipoR2 tiene mayor afinidad por la adiponectina completa (full-lenght), mientras que el AdipoR1 tiene mayor afinidad por una forma corta de adiponectina generada por proteólisis. Los niveles plasmáticos de adiponectina guardan una fuerte relación inversa con el peso corporal y una fuerte relación directa con la sensibilidad a la insulina, como se ha demostrado en estudios epidemiológicos y en estudios con intervención farmacológica para mejorar la sensibilidad a la insulina (41-44).
La adiponectina se caracteriza por poseer efectos biológicos que se podrían llamar "protectores" o "antiaterogénicos" (41-44):
Reduce la producción hepática de glucosa.
Estimula la beta-oxidación de ácidos grasos en hígado.
Inhibe la adhesión de monocitos al endotelio vascular.
Inhibe la expresión de receptores basurero ("scavenger") de LDL en los macrófagos.
Inhibe la proliferación y migración de células musculares lisas en la pared arterial.
Incrementa la fosforilación del receptor de insulina, y por ende todos los demás efectos insulínicos.
Como hemos pretendido hacer notar, los mecanismos celulares y moleculares de la acción insulínica y la resistencia a la insulina, involucrados en el desarrollo y progresión de la diabetes tipo 2 son múltiples, complejos, intrincados y sólo parcialmente conocidos.
Avanzar en la comprensión de éstos mecanismos, así como identificar potenciales blancos terapéuticos en niveles diferentes a los tradicionales, repercutirá en el tratamiento y pronóstico de los pacientes con diabetes tipo 2, así como en la calidad de vida de la población en general; dada la importancia de la diabetes tipo 2 a nivel de salud pública.
Referencias
1. Le Roith D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001;243:588-597. [ Links ]
2. Sierra ID: Estados metabólicos nutricionales. En: Sierra ID. Metabolismo de los carbohidratos y su importancia clínica. 2ª Ed. Bogotá: Editorial Kimpres, 1999. [ Links ]
3. Flakoll PJ, Carlson MG, Cherrington AD. Acción fisiológica de la insulina. En: Le Roith D, Taylor SI, Olefsky JM, editores. Diabetes Mellitus, Fundamentos y Clínica. 2ª ed. México: Mc Graw Hill, 2003. [ Links ]
4. Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994;43:1066-84. [ Links ]
5. Ullrich A, Bell JR, Chen EY. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985;313:756-61. [ Links ]
6. Shoelson SE, White MF, Kahn CR. Tryptic activation of the insulin receptor. Proteolytic truncation of the alfa-subunit releases the beta-subunit from inhibitory control. J Biol Chem 1988;263:4852-60. [ Links ]
7. Pirola L, Johnston AM, Van Obberghen E. Modulation of insulin action. Diabetologia 2004;47:170-84 [ Links ]
8. Giovannone B, Scaldaferri ML, Federici M et al. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res Rev 2000;16:434-41. [ Links ]
9. Myers MG, White MF. The new elements of insulin signaling. Insulin receptor substrate-1 and proteins with SH2 domains. Diabetes 1993;42:643-650. [ Links ]
10. Araki E, Lipes MA, Patti ME. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994;372:186-90. [ Links ]
11. Tamemoto H, Kadowaki T, Tobe K. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 1994;372:182-6. [ Links ]
12. Withers DJ, Gutierrez JS, Towery H et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900-4. [ Links ]
13. Pons S, Asano T, Glasheen E et al. The structure and function of p55PIK reveals a new regulatory subunit for phosphatidylinositol 3-kinase. Mol Cell Biol 1995;15:4453-65. [ Links ]
14. Kohjiro Ueki, David A. Fruman et al. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-Kinase regulates cell signaling and survival. Mol Cell Biol 2002;22:965-77. [ Links ]
15. Currie RA, Walker KS, Gray A et al. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase. Biochem J 1999;337:575-83. [ Links ]
16. Wada T, Sasaoka T, Funaki M et al. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5'-phosphatase catalytic activity. Mol Cell Biol 2001;21:1633-46. [ Links ]
17. Yang C, Watson RT, Elmendorf JS et al. Calmodulin antagonists inhibit insulin-stimulated GLUT4 (glucose transporter 4) translocation by preventing the formation of phosphatidylinositol 3,4,5-trisphosphate in 3T3L1 adipocytes. Mol Endocrinol 2000;14:317-26. [ Links ]
18. Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J 1998 ;333:471-90. [ Links ]
19. Okada T, Kawano Y, Sakakibara T et al. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem 1994;269:3568-73. [ Links ]
20.Skolnik EY, Lee CH, Batzer A et al. The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implications for insulin control of ras signalling. EMBO J 1993;12:1929-360. [ Links ]
21. Skolnik EY, Batzer A, Li N et al. The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science 1993;260:1953-5. [ Links ]
22. Van Dam EM, Govers R, James DE. Akt activation is required at a late stage of insulin-induced GLUT4 translocation to the plasma membrane. Mol Endocrinol 2005;Publicación electrónica previa a la impresa. [ Links ]
23. Sakamoto K, Aschenbach WG, Hirshman MF et al. Akt signaling in skeletal muscle: regulation by exercise and passive stretch. Am J Physiol Endocrinol Metab 2003;285:E1081-8. [ Links ]
24. Wojtaszewski JF, Nielsen P, Kiens B et al. Regulation of glycogen synthase kinase-3 in human skeletal muscle: effects of food intake and bicycle exercise. Diabetes 2001;50:265-9. [ Links ]
25. Bhandari BK, Feliers D, Duraisamy S et al. Insulin regulation of protein translation repressor 4E-BP1, an eIF4E-binding protein, in renal epithelial cells. Kidney Int 2001;59:866-75. [ Links ]
26. Kellerer M, Coghlan M, Capp E et al. Mechanism of insulin receptor kinase inhibition in non-insulin-dependent diabetes mellitus patients. Phosphorylation of serine 1327 or threonine 1348 is unaltered. J Clin Invest 1995;96:6-11. [ Links ]
27. Grasso G, Frittitta L, Anello M et al. Insulin receptor tyrosine-kinase activity is altered in both muscle and adipose tissue from non-obese normoglycaemic insulin-resistant subjects. Diabetologia 1995;38:55-61. [ Links ]
28. Acilli D, Nakae J, Flier JS. Receptor de insulina. En: Le Roith D, Taylor SI, Olefsky JM, eds. Diabetes Mellitus, Fundamentos y Clínica. 2ª edición. México: Mc Graw Hill, 2003. [ Links ]
29. Newell AM. Genetics for targeting disease prevention: diabetes. Prim Care 2004;31:743-66 . [ Links ]
30. Freidenberg GR, Reichart D, Olefsky JMet al. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin-dependent diabetes mellitus. Effect of weight loss. J Clin Invest 1988;82:1398-406. [ Links ]
31. Hotamisligil GS. Molecular mechanisms of insulin resistance and the role of the adipocyte. Int J Obes 2000;24(suppl 4):S23-S27. [ Links ]
32. Fasshauer M, Paschke R. Regulation of adipocytokines and insulin resistance. Diabetologia 2003;46:15941603. [ Links ]
33. Kershaw EE, Flier JS. Adipose Tissue as an Endocrine Organ. J Clin Endocrinol Metab 2004;89:2548-56. [ Links ]
34. Garg A. Regional Adiposity and Insulin Resistance. J Clin Endocrinol Metab 2004;89:4206-4210. [ Links ]
35. Wolf G. Insulin resistance and obesity: resistin, a hormone secreted by adipose tissue. Nutr Rev 2004 Oct;62:389-94. [ Links ]
36. Schmitz-Peiffer C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann N Y Acad Sci 2002;967:146-57. [ Links ]
37. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7. [ Links ]
38. Standaert ML, Ortmeyer HK, Sajan MP et al. Skeletal muscle insulin resistance in obesity-associated type 2 diabetes in monkeys is linked to a defect in insulin activation of protein kinase C-zeta/lambda/iota. Diabetes 2002;51:2936-43. [ Links ]
39. Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor. Cytokine Growth Factor Rev 2003;14:447-455. [ Links ]
40. Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord 2003;27(Suppl 3):S53-S55. [ Links ]
41. Tsao TS, Lodish HF, Fruebis J. ACRP30, a new hormone controlling fat and glucose metabolism. Eur J Pharmacol 2002;440:213-221. [ Links ]
42. Matsuzawa Y, Funahashi T, Kihara S et al. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol 2004;24:2933. [ Links ]
43. Chandran M, Phillips SA, Ciaraldi T et al. Adiponectin: more than just another fat cell hormone? Diabetes Care 2003;26:2442-2450. [ Links ]
44. Diez JJ, Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol 2003;148:293-300. [ Links ]
