










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.61 no.3 Bogotá July/Sept. 2013
Tumor neuroectodérmico primitivo de la pared torácica: tumor de Askin en niños
Primitive neuroectodermal tumor of the chest wall: Askin tumor in children
David Bravo-Linares¹ • Katherine Hernández-Verbel MD² • Adriana Linares-Ballesteros MD² • Lina Jaramillo-Barberi MD3,4 • Mizrahim Méndez-Manchola MD4,5 • Fabio Restrepo-Ángel MD2,4 • Isabel Sarmiento-Urbina MD2 • Fernando Fierro-Ávila MD5
1 Universidad Nacional de Colombia. Bogotá.
2 Departamento de Pediatría, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá.
3 Departamento de Patología, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá.
4 Fundación Hospital de La Misericordia. Bogotá.
5 Departamento de Cirugía, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá.
Correspondencia: Adriana Linares Ballesteros, Avenida Caracas No. 1-67. 4º piso. Departamento de Pediatría. Bogotá, Colombia. Teléfono: 57 1 3373842. Correo electrónico: talinaresb@unal.edu.co
Recibido: 28/01/2013 / Aceptado: 19/07/2013
Resumen
Antecedentes. El tumor neuroectodérmico primitivo (TNEP) de la pared torácica o Tumor de Askin, es un tumor maligno clasificado dentro de los TNEP periféricos y perteneciente a los tumores de la familia del sarcoma de Ewing (FSE). Este tipo de neoplasia de la región toracopulmonar es muy poco frecuente y suele presentarse con mayor frecuencia en la población pediátrica.
Objetivo. Describir algunas características de pacientes con Tumor de Askin de la Fundación Hospital La Misericordia.
Materiales y métodos. Se realizó un análisis retrospectivo de los casos atendidos en una sola institución, Fundación Hospital de la Misericordia, en un lapso de 16 años.
Resultados. Se encontraron 8 pacientes, 7 varones, 4 con metástasis al diagnóstico, 2 en pulmón y 2 en hueso, a 2 se les hizo resección quirúrgica completa del tumor al diagnóstico, los demás pacientes fueron llevados a biopsia y quimioterapia citoreductora previa a la cirugía para resección tumoral. Todos recibieron quimioterapia con protocolos para Sarcoma de Ewing y radioterapia en 6 casos. A la fecha están vivos 5 pacientes (62%) con una mediana de seguimiento de 32 meses; 3 de ellos tenían metástasis en el diagnóstico. De los 3 pacientes muertos, 2 no tenían metástasis en el diagnóstico, ambos recayeron y fallecieron por progresión de enfermedad; el otro abandonó el tratamiento a los 6 meses del diagnóstico. Dos pacientes en recaída recibieron quimioterapia de 2 líneas a altas dosis con rescate autólogo, uno está vivo y el otro falleció por progresión de la enfermedad.
Conclusión. Se describieron las características clínicas, de laboratorio, diagnóstico, tratamiento y pronóstico de este grupo de pacientes.
Palabras clave: Tumor Neuroectodérmico Melanótico, Tumores Neuroectodérmicos Periféricos Primitivos, Niño (DeCS).
Summary
Background. The primitive neuroectodermal tumor (TNEP) of the chest wall, or Askin tumor, is a malignant neoplasia classified within the Ewing family of tumours (FSE). This particular type of malignancy of the chest wall is rare and usually appears in the pediatric population.
Objective. Describe some characteristics of patients with Askin tumor of the Fundacion Hospital de La Misericordia.
Materials and methods. A retrospective analysis of cases treated at a single institution, Fundación Hospital de la Misericordia, in a 16 years period was performed.
Results. 8 patients were found, 7 boys, 4 with metastases at the diagnosis, 2 in lungs and 2 in bone, 2 underwent surgery at diagnosis. For the other 6 patients surgical resection was done after chemotherapy. All patients received chemotherapy and 6 received radiotherapy. To date, 5 patients are alive (62 %) with a median follow up of 32 months; 3 of them had metastases at diagnosis. 3 patients died, 2 of which had no metastases in the diagnosis, both relapsed and died of disease progression, the other abandoned treatment at 6 months after diagnosis. Other 2 patients that relapsed received chemotherapy plus autologous transplant, one is alive and the other one died due to disease progression.
Conclusion. Clinical, laboratory, diagnosis, treatment and prognosis characteristics of this patient group were described.
Key Words: Neuroectodermal Tumor, Melanotic; Neuroectodermal Tumors, Primitive, Peripheral; Child (MeSH).
Introducción
El primer reporte del tumor neuroectodérmico primitivo de la pared torácica fue hecho por Askin y Rosai en 1979 cuando describieron 20 pacientes, 75% de ellos mujeres, en promedio de 14,5 años de edad, con neoplasias originadas en los tejidos blandos de la pared torácica y tejido pulmonar periférico con un comportamiento agresivo (1). Los tumores neuroectodérmicos primitivos (TNEP) derivan de células pluripotenciales neuroectodérmicas de la cresta neural y pertenecen al grupo de tumores de células pequeñas redondas y azules que se caracterizan por un comportamiento agresivo, con manifestaciones clínicas diversas que dependen de su localización anatómica (2,3). Batsakis en 1996 dividió los TNEPs en 3 grupos según su tejido de origen: meduloblastoma cuando estaba en el sistema nervioso central, neuroblastoma si era del sistema nervioso autónomo y TNEP periféricos (pTNEPs) si se encontraban en tejidos blandos (4). En la actualidad se continúan manejando como sinónimos nombres tan diversos como neuroepitelioma periférico, neuroblastoma periférico y tumor de Askin. Según la definición actual de la OMS, el Sarcoma de Ewing y el TNEP corresponden a un sarcoma de células redondas que muestra grados diversos de diferenciación. Se conocen entonces como miembros de los tumores de la familia del sarcoma de Ewing (FSE), una familia de neoplasias que comparten anormalidades genéticas que comprometen los cromosomas 11 y 22, caracterizados por una translocación recurrente t(11;22)(q24;q12) o variantes de esta translocación en otros cromosomas que comprometen el oncogén EWS. Los rearreglos del gen EWS (22q12) y la fusión con otros factores de trascripción llevan a la activación de proteínas de transcripción aberrantes y a la pérdida de la regulación normal del crecimiento en esta familia de neoplasias (5,18). El término sarcoma de Ewing se aplica más a los tumores primarios de la medula del hueso, mientras que el de tumor neuroectodérmico primitivo periférico se usa en las neoplasias originadas de tejidos blandos (2).
Este reporte describe la experiencia con pacientes diagnosticados con tumor de Askin en un período de 16 años en el servicio de oncohematología pediátrica de la Fundación Hospital de la Misericordia, adicionalmente se hace una revisión del tema de esta patología poco común y poco registrada en la literatura nacional y mundial.
Materiales y métodos
Se realizó un análisis retrospectivo de los casos atendidos en el servicio de oncohematología pediátrica de la Fundación Hospital de la Misericordia en un lapso de 16 años entre 1995 y 2011.
Resultados
Se encontraron 8 pacientes menores de 18 años, el promedio de edad fue de 9 años (DE 2.3), 7 niños y una niña, todos los pacientes tuvieron tumores originados en arcos costales con extensión al tejido blando; el período de seguimiento estuvo entre 8 y 84 meses (mediana 32 meses RIQ 9-84) (Tablas 1 y 2). Los pacientes recibieron terapia multimodal con cirugía, quimioterapia y radioterapia. Dos pacientes fueron llevados a cirugía con resección completa del tumor al diagnóstico, 6 pacientes se trataron posteriores a biopsia de la lesión, con quimioterapia seguida de resección quirúrgica del tumor primario. Seis pacientes recibieron radioterapia. La morfología de los tumores reportó en todos ellos un tumor de células redondas pequeñas y azules con necrosis frecuente y el diagnóstico de TNEP se confirmó por Inmunohistoquímica, observando expresión de CD99 en todos los casos con el característico patrón de membrana y en proporciones diversas para marcadores neurales como enolasa neuroespecífica (NSE), CD57 y synaptofisina.
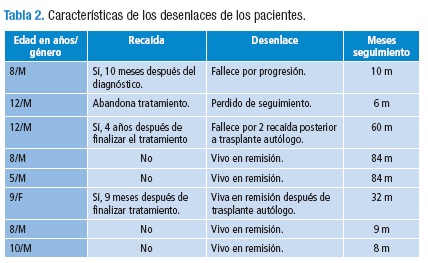
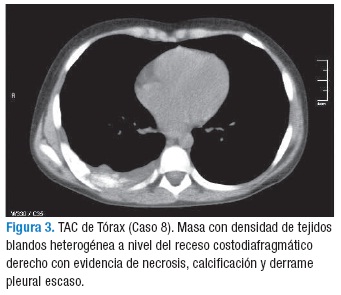
Los hallazgos radiológicos fueron: masa heterogénea con mayor frecuencia del hemitórax izquierdo (5/8), con imágenes líticas o necrosis y extensión a los tejidos blandos (4/8), derrame pleural ipsilateral, desplazamiento del cardiomediastino y colapso pulmonar (3/8). Sólo un caso mostró calcificaciones en el tumor. La mayoría tenía masa de gran volumen al momento de la consulta.
De los 8 pacientes, 5 (62%) están vivos y sin evidencia de enfermedad. 3 de ellos tenían metástasis en el diagnóstico, 1 recayó y se consolidó con trasplante. De los 3 pacientes muertos uno abandonó el tratamiento a los 6 meses, los otros 2 no tenían metástasis en el diagnóstico, ambos recayeron, uno falleció en progresión de la enfermedad y el otro por segunda recaída
Discusión
El tumor de Askin hace parte de los tumores de la familia del sarcoma de Ewing, es un tumor extremadamente agresivo y raro, ocurre con mayor frecuencia en niños y adolescentes, la incidencia es del 27% en la primera década de vida, del 64% en la segunda y hasta un 9% en la tercera década (6). En nuestra serie de casos encontramos 6/8 casos en la primera década de la vida (promedio 9 años).
No se conoce la incidencia exacta del tumor de Askin, se dice que aproximadamente el 15% de los tumores de la familia del Sarcoma de Ewing corresponden a tumores de la pared torácica (6-8). Los tumores derivados de la pleura y la pared torácica como grupo, son poco frecuentes en pediatría. En la revisión de los tumores sólidos en 22 años de la institución Fundación Hospital de la Misericordia se encontró que los tumores de la pared del tórax fueron 1 % del total de los tumores malignos; dentro de este grupo el tumor de Askin fue el más frecuente seguido por el blastoma pleuropulmonar. La incidencia de los FSE es de 1 a 3 personas por millón de habitantes al año, los pocos estudios han mostrado una mayor frecuencia en varones, con una relación hombre mujer 1.5:1, tal como lo vimos en esta serie donde encontramos marcado predominio en el género masculino (7/8). La literatura dice que al momento del diagnóstico un 80% de los pacientes presentan enfermedad localizada, y 20-25% tienen metástasis (9,10). En nuestro trabajo infortunadamente encontramos que el 50% de los pacientes tenían metástasis en el momento del diagnóstico; las causas podrían ser varias y debemos considerar entre otras la demora en la consulta, falta de sospecha de tumor o que se trate de una enfermedad más agresiva.
Tal y como se encuentra en las publicaciones encontramos que en nuestros pacientes la sintomatología previa a la consulta presentó una mediana de 4,5 meses (1,9). Clínicamente los pacientes se manifiestan con disnea, tos, pérdida de peso, fiebre y aumento en el tamaño o sensación de masa asociado a dolor, siendo este último el síntoma que, con mayor frecuencia, aparece tempranamente. En esta serie de casos encontramos que el síntoma más frecuente fue la presencia de dolor asociado a masa (7/8), disnea (4/8), fiebre (4/8), pérdida de peso (4/8) y tos (2/8); como en muchos casos de tumores en niños, los pacientes relacionan la aparición de la masa con antecedente de trauma previo en la zona de la lesión, 3/8 de nuestros pacientes así lo referían (1,9).
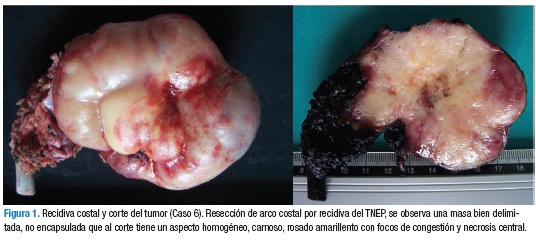
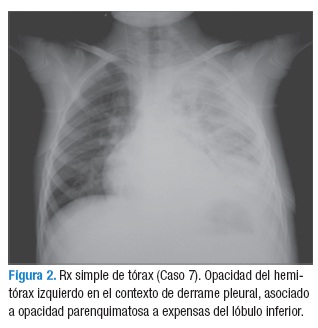
El tumor de Askin compromete no sólo la pared torácica sino que invade los tejidos blandos adyacentes, periostio y parénquima pulmonar por extensión directa (1). Su diagnóstico se basa en estudios imagenológicos y se confirma con el estudio de histopatología en biopsia o en la resección quirúrgica. Los estudios imagenológicos para evaluación del tumor primario incluyen una radiografía de tórax en proyección PA y lateral (Figura 2), TAC y RMN de tórax (Figura 3). Los principales hallazgos radiológicos muestran derrame pleural unilateral o bilateral con engrosamiento pleural, masa en la pared torácica unilateral de características heterogéneas que se extiende a los músculos de la pared torácica, es poco frecuente encontrar calcificaciones. Adicionalmente, hay invasión del parénquima pulmonar adyacente y destrucción costal y necrosis. En esta serie de casos se encontró masa heterogénea en (5/8) en hemitórax izquierdo y (3/8) en hemitórax derecho, derrame pleural (4/8), metástasis pulmonar (2/8) y metástasis óseas (2/8) pacientes, a uno se le realizó gammagrafía ósea, evidenciando hipercaptación en séptimo arco costal derecho; no se encontró infiltración tumoral de medula ósea. Los estudios imagenológicos de extensión comprenden TAC abdominopélvica, gammagrafía ósea y tomografía por emisión de positrones (PET). En estados avanzados, se encuentran metástasis pulmonares, siendo éstas, junto con las metástasis de hueso y médula ósea, las más frecuentes (1,9,11,12). Otros procedimientos complementarios incluyen el hemograma, lactato deshidrogenasa, catecolaminas en orina, velocidad de sedimentación globular, función hepática y renal, aspirado y biopsia de médula ósea como estudios de extensión.
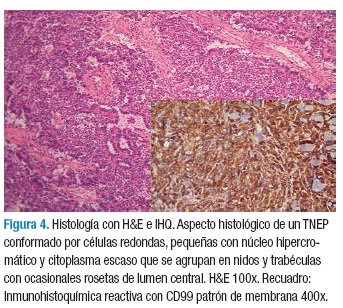
El diagnóstico confirmatorio se establece con el estudio histológico, donde se observa un tumor de células pequeñas, hipercromáticas, de núcleo oval o redondo con citoplasma mínimo que se agrupan en nidos y trabéculas formando ocasionalmente rosetas con un lumen central (13,14). Es frecuente observar necrosis, cariorrexis y alto índice mitótico. El diagnóstico definitivo sólo es posible hacerlo con estudios de inmunohistoquímica. Los FSE/TNEP presentan niveles elevados de un antígeno, el cual es codificado por el gen MIC-2, el resultado de este gen es una glicoproteína de superficie CD99 (p30/32MIC2); los anticuerpos monoclonales para esta glicoproteína, se muestran positivos en el 95-100 % de los casos, siendo entonces el principal marcador diagnóstico. Se puede encontrar reactividad igualmente para vimentina y marcadores neurales como la enolasa neuronal específica (NSE), cromogranina, S-100, Leu7 (CD57), CD75 y synaptofisina.
El diagnóstico del tumor de Askin ha sido en gran medida un proceso de exclusión para diferenciarlo de otros tumores de la familia del sarcoma de Ewing (5,6,15). En este reporte de casos, todos los estudios de inmunohistoquimica fueron positivos para CD99 y también para otros inmunomarcadores como NSA, vimentina y synaptofisina, que fueron positivos en la mayoría de los pacientes (Figura 4).
Con los avances en el tratamiento, el pronóstico ha mejorado y se han identificado varios factores de riesgo para la estratificación de los pacientes en diferentes grupos. Los criterios más utilizados para esta estratificación son el tamaño del tumor, la presencia o ausencia de metástasis y la respuesta histológica después de la inducción de la quimioterapia.
Los pacientes se dividen en el momento del diagnóstico como enfermedad localizada o metastásica y se debe considerar como una enfermedad sistémica que requiere, por tanto, un control local basado en resección quirúrgica siempre que sea posible. Adicionalmente, se ha usado radioterapia y quimioterapia intensiva combinada. El tratamiento con quimioterapia pretende obtener una respuesta adecuada, reducir la toxicidad, evitar la enfermedad metastásica, la recurrencia del tumor y poder tratar el tumor residual después de la resección quirúrgica (16). Los pacientes de esta serie, 2/8 fueron llevados a resección quirúrgica del tumor al diagnóstico, todos recibieron quimioterapia, y 6/8 recibieron radioterapia; 6/8 fueron llevados a resección quirúrgica después de 2 a 4 ciclos de quimioterapia. En 2 casos se practicó trasplante autólogo de medula ósea por recaída de la enfermedad después de alcanzar buena respuesta y entrando en una segunda remisión completa (9,17).
El pronóstico del tumor de Askin está dado por el compromiso sistémico de cada caso; los pacientes con compromiso local tienen una tasa de supervivencia hasta del 70 %, los que se presentan con metástasis pulmonares tienen una supervivencia alrededor del 40 %, los que tienen enfermedad diseminada ya sea en hueso, médula ósea o recurrencia local del tumor, la tasa de supervivencia es cerca del 20 % a 5 años (9,10). En este grupo de pacientes, a la fecha, con una mediana de seguimiento de 32 meses, están vivos 5 pacientes, 3 de ellos con metástasis al diagnóstico.
Financiación
Ninguna declarada por los autores.
Conflictos de interés
Ninguno declarado por los autores.
Agradecimientos
Ninguno declarado por los autores.
Referencias
1. Askin F, Rosai J, Sibley R, Dehner L, Mcalister W. Malignant small cell tumor of the thoracopulmonary region in childhood: a distinctive clinico-pathologic entity of uncertain histogenesis. Cancer. 1979;43:2438-51. [ Links ]
2. La Quaglia MP. Chest wall tumors in childhood and adolescence. Semin Pediatr Surg. 2008;17:173-180. [ Links ]
3. Huang M, Lucas K. Current Therapeutic Approaches in Metastatic and Recurrent Ewing Sarcoma. Sarcoma. 2011;1-5. [ Links ]
4. Batsakis J, Mackay B, El-Naggar A. Ewing's sarcoma and peripheral primitive neuroectodermal tumor: an interim report. Ann Otol Rhinol Laryngol. 1996;838-43. [ Links ]
5. De Alava E, Gerald W. Molecular Biology of the Ewing's Sarcoma/Primitive Neuroectodermal Tumor Family. J Clin Oncol. 2000;18:204-13. [ Links ]
6. Bernstein M, Kovar H, Paulussen M, Lor R, Schuck A, Teot L, et al. Ewing's Sarcoma Family of Tumors: Current Management. Oncologist. 2006;11:503-19. [ Links ]
7. Windfuhr J. Primitive neuroectodermal tumor of the head and neck: incidence, diagnosis, and management. Ann Otol Rhinol Laryngol. 2004;7:533-43. [ Links ]
8. Laskar S, Nair C, Mallik S, Bahl G, Pai S, Shet T, el al. Prognostic factors and outcome in Askin-Rosai tumor: a review of 104 patients. Int J Radiat Oncol Biol Phys. 2011;79:202-7. [ Links ]
9. Potratz J, Dirksen U, Jurgens H, Craft A. Ewing Sarcoma: Clinical State-of-the-Art. Pediatr Hematol Oncol. 2012;29:1-11. [ Links ]
10. Paulussen M, Bielack S, Jürgens H, Casali P. Ewing's sarcoma of the bone: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20 Supl 4:14-142. [ Links ]
11. Winer-Muram H, Kauffman W, Gronemeyer S, Jennings S. Primitive Neuroectodermal Tumors of the Chest Wall (Askin Tumors): CT and MR Findings. AJR. 1993;161:265-8. [ Links ]
12. Hari S, Jajn T, Thulkar S, Bakhshi S. Imaging features of peripheral primitive neuroectodermal tumours. BJR. 2008;81:975-83. [ Links ]
13. Moreno J, Rey C. Metástasis de origen desconocido en tumores óseos. Glaxo Wellcome. 1998;173-83. [ Links ]
14. Davidoff A, Fernandez I, Santana V. The role of neoadjuvant chemotherapy in children with malignant solid tumors. Semin Pediatr Surg. 2012;21:88-99 [ Links ]
15. May W, Lessnick S, Braun B, Klemsz M. The Ewing's Sarcoma EWS/FLI-1 Fusion Gene Encodes a More Potent Transcriptional Activator and Is a More Powerful Transforming Gene than FLI-1. Mol Cell Biol. 1993;13:7393-98. [ Links ]
16. Dang N, Siegel S, Duncan J. Malignant Chest Wall Tumors in Children and Young Adults. J Pediatr Surg. 1999;34:1773-8. [ Links ]
17. Indelicato D, Keole SR, et al. Chest wall ewing sarcoma family of tumors: long-term outcomes. Int. J. Radiation Oncology Biol. Phys. 2011;81:158-166. [ Links ]
18. Folpe A, Goldblum J, Rubin B, Shehata B, Liu W. Morphologic and immunophenotypic diversity in Ewing family tumors: A study of 66 genetically confirmed cases. Am J Surg Pathol. 2005;29:1025-33. [ Links ]
