










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista MVZ Córdoba
Print version ISSN 0122-0268On-line version ISSN 1909-0544
Rev.MVZ Cordoba vol.10 no.1 Córdoba Jan./June 2005
REVISIÓN DE LITERATURA
PATOGENESIS DE Listeria monocytogenes, MICROORGANISMO ZOONOTICO EMERGENTE.
PATHOGENESIS OF Listeria monocytogenes, MICROORGANISM ZOONOTIC EMERGENT
Kirvis Torres1, Sara Sierra1, Raúl Poutou1*, Ana Carrascal1, Marcela Mercado2.
1Laboratorio de Microbiología de Alimentos, Laboratorio de Biotecnología Aplicada, Grupo de Biotecnología Ambiental e Industrial.
2Grupo de Enfermedades Infecciosas. Depto. Microbiología, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá, D.C.
*Correspondencia:rp000274@javeriana.edu.co
RESUMEN
Listeria monocytogenes además de ser un paradigma para la investigación inmunológica se ha convertido en sistema modelo apropiado para el análisis de los mecanismos moleculares del parasitismo intracelular de otras bacterias. Investigadores en el área de la inmunología se interesaron en este microorganismo cuando se reconoció el riesgo que representaba para la salud pública y la seguridad en la industria de alimentos. Desde mediados de los años 80’s se ha investigado la biología molecular de los marcadores de virulencia de este microorganismo, la biología celular de las interacciones de los marcadores de virulencia con los receptores de la célula hospedero, el citoesqueleto, las vías de transducción de señales y los mecanismos de inmunidad mediada por células del hospedero. El propósito de esta revisión es describir algunas características taxonómicas y filogenéticas de Listeria monocytogenes, la incidencia humana y animal de varios serotipos, la fisiopatología de la infección, modelos animales y de cultivo celular utilizados para estudios de virulencia, las poblaciones de riesgo, manifestaciones clínicas de listeriosis humana y animal, el tratamiento, la organización genética y evolución de los determinantes de virulencia, los mecanismos empleados para interactuar con la célula hospedera, y los mecanismos para escapar de los procesos de muerte celular y pasar de una célula infectada a otra. La información recopilada resulta de gran importancia para el personal de salud, industria, consumidores y población de riesgo; razón por la cual Listeria monocytogenes es un patógeno que representa una amenaza para la salud pública mundial.
Palabras clave: Patogénesis, Listeria monocytogenes, resistencia antimicrobiana, biología molecular, serotipo
ABSTRACT
Listeria monocytogenes in addition to being a paradigm for the immunological investigation has become in an appropriate model system to the analysis of the molecular mechanisms of the intracellular parasitism of other bacteria strains. Inmunologyst were interested in this microorganism when was recognized as a risky organism for public health and the food industry security. From mid of the 80’s scientists have researched the molecular biology of virulence markers of this microorganism, the cellular biology of the interactions of these markers with the receptors of the host cell, the cytoskeleton, the transduction signals routes and the mechanisms of immunity mediated by the host cells. The intention of this review is to describe some taxonomic and phylogenetics characteristics of Listeria monocytogenes, the human and animal incidence of several serotypes, the physiopathology of the infection, animals models and cellular culture employed for virulence studies, the risk populations, clinical manifestations of human and animal listeriosis, the treatment, the genetic organization and evolution de the virulence markers, the mechanisms used to interact with the host cell, the mechanism to escape from the cellular death processes and to pass through an infected cell to another one. The compiled information is from great importance for the health personnel, consumers and risk population; reason for which Listeria monocytogenes it is a pathogen that represents a threat for world-wide the public health.
Key words: Pathogenesis, Listeria monocytogenes, antimicrobial resistance, molecular biology, serotype.
INTRODUCCIÓN
La listeriosis es una enfermedad bacteriana invasiva, producida por Listeria monocytogenes; cocobacilo Gram-positivo psicrótrofo, móvil, no esporulado, anaerobio facultativo, patógeno de origen alimentario en humanos y animales con una amplia distribución en la naturaleza. La contaminación alimentaria, en epidemias o en casos esporádicos, en poblaciones inmunosuprimidas, constituyen los dos factores fundamentales para la presentación de la enfermedad. La amplia distribución de Listeria monocytogenes se debe a la capacidad de sobrevivir durante períodos de tiempo prolongados en diferentes medios. Por consiguiente, los alimentos se pueden contaminar en cualquier eslabón de la cadena productiva y en el almacenamiento en frío.
El parasitismo intracelular de L. monocytogenes es considerado un modelo importante para la investigación de los mecanismos moleculares de patogénesis intracelular bacterianos (Shen et al., 1998). Conceptos importantes como la inhabilidad de los anticuerpos para proteger contra infecciones producidas por patógenos intracelulares, la importancia de macrófagos activados en la eliminación de parásitos intracelulares, y el papel que juega los linfocitos T en la inmunidad celular inmediata fueron establecidos bajo los estudios con el modelo murino de listeriosis (North 1978).
CARÁCTERÍSTICAS FILOGENÉTICAS
El género Listeria está constituido por seis especies, Listeria innocua, Listeria weishimeri, Listeria seeligeri, Listeria ivanovii, Listeria grayi, Listeria murrayi. Únicamente dos especies de este género son patogénicas: L. monocytogenes asociada con infección en humanos y animales y Listeria ivanovii asociada únicamente con infección en animales (Seeliger 1986).
El genoma de L. monocytogenes fue secuenciado recientemente y posee un cromosoma circular de 2.944.528pb con un promedio de G + C de 39%. Se han identificado 2853 genes, sin embargo al 35.3% de estos no se les conoce función. De otro lado, L. innocua posee un único cromosoma circular de 3.011.209pb con un contenido promedio de G + C de 37%. Dentro del género Listeria, estas dos especies presentan alto grado de homología en la secuencia del RNA ribosomal 16S (RNAr 16S) siendo las de mayor cercanía taxonómica (Figura 1) (Von Both et al., 1999).
La habilidad de Listeria sp., para colonizar y crecer en un amplio rango de ecosistemas se correlaciona con la presencia de 331 genes que codifican para diferentes proteínas de transporte. El número de componentes reguladores del genoma de Listeria monocytogenes es similar a otras bacterias, tales como Bacillus subtilis, Escherichia coli, Clostridium spp., Staphylococcus spp., Streptococcus spp., Lactobacillus spp., Bronchothrix spp., y superior a M. tuberculosis y Neisseria meningitidis. El análisis de la secuencia de los genomas de L. monocytogenes y L. innocua revelan una relación filogenética con B. subtilis, lo que sugiere un origen común para las tres especies.
SEROTIPIFICACIÓN
La heterogeneidad en la virulencia de L. monocytogenes ha sido observada en estudios in vivo (ratón) y en estudios in vitro (cultivos celulares); pero la correlación entre el nivel de virulencia y el origen o tipo de cepa no ha sido establecida (Brosch et al., 1993). Sin embargo, se han encontrado diferencias significativas entre la virulencia de cepas de origen clínico y de alimento, siendo las primeras las que presentan dosis letales más bajas (Norrung 2000). Basados en los antígenos somático (O) y flagelar (H), existen hasta el momento, 13 serovariedades de L. monocytogenes. Sin embargo, tres de ellas (½a, ½b y 4b) han sido aisladas en más del 90% de los casos de listeriosis humana y animal (Low et al., 1993). Otras serovariedades, como el ½c, ha sido encontrada como contaminantes de alimentos (Espaze et al., 1991). Algunas de estas serovariedades son compartidas por L. innocua y por L. seeligeri (Menudier et al., 1991). L. innocua está representada sólo por tres serovariedades y es considerada una variante no patógena de L. monocytogenes (Tabla 1).
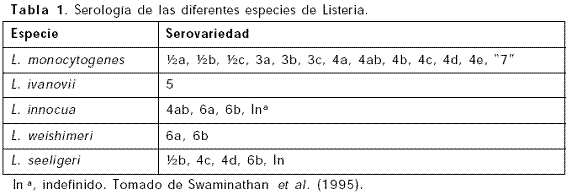
Entre las serovariedades asociadas a listeriosis, las cepas 4b causan más del 50% de los casos en todo el mundo, pero las cepas de grupo antigénico ½ (½a, ½b y ½c) predominan en los aislamientos a partir de alimentos (Rocourt 1994a; Rocourt 1994b); esto sugiere que las cepas serotipo 4b están más adaptadas a tejidos de hospedero mamífero que las cepas del serogrupo ½ (Swaminathan et al., 1995).
Un grupo de cepas relacionadas fenotípica y genómicamente de la serovariedad 4b fueron encontrados como responsables de los brotes más importantes de listeriosis humana transmitidos por alimentos en California en 1985 (Linnan et al., 1988), Suiza entre 1983 y 1987 (Bille 1990), Dinamarca entre 1985 y 1987 (Samuelsson et al., 1990) y Francia en 1992 (Goulet et al., 1993), soportando la idea de que la patogenia está asociada a ciertos clones de L. monocytogenes (Jacquet et al., 1995).
Wiedman en 1997, encontró una correlación entre las tres serovariedades relacionadas evolutivamente, que permitió agrupar los aislamientos de L. monocytogenes (Rasmussen et al., 1995; Wiedmann et al., 1997) y determinar el potencial patogénico para humanos y animales. Así, un grupo de cepas (serovariedades ½b y 4b) fue aislado de epidemias de origen alimentario en humanos y de casos esporádicos en humanos y animales, otro grupo de cepas (serovariedades ½a, ½c y 3a) fueron aisladas únicamente de casos esporádicos en humanos y animales y un tercer grupo (serovariedad 4b) aislado de casos esporádicos en animales.
Aunque Gray y Killinger en 1966 (Gray 1966) indicaron que las serovariedades de Listeria no están relacionadas con el hospedero, el proceso patológico y el origen geográfico; el posible tropismo de ciertos clones de L. monocytogenes por el humano pueden explicar, las observaciones hechas en Scotland (Reino Unido), donde en el mismo período de tiempo la serovariedad 4b fue aislada de casos humanos, mientras los casos de rumiantes estuvieron relacionados con la serovariedad ½a (Low et al., 1993).
Existen diferencias en el tropismo patogénico entre cepas de L. monocytogenes. En humanos, por ejemplo, las cepas serovariedad 4b han sido aisladas de fetos más que de casos no asociados con el embarazo (McLauchlin 1990). En ovejas, las dos formas clínicas de infección por L. monocytogenes, meningoencefalitis y aborto, no ocurren simultáneamente en el mismo rebaño (Vázquez-Boland et al., 1996).
En un estudio realizado por el Laboratorio de Salud Pública del Distrito Capital (Bogotá, Colombia) en muestras de derivados cárnicos tipo jamón, procedentes de diferentes hospitales del Distrito, entre Enero y Agosto del 2002, se aislaron las serovariedades 4b y ½b, siendo la serovariedad 4b la predominante (Comunicación personal Herber Vera, Referente de Ambiente del Distrito).
FISIOPATOLOGIA DE LA INFECCIÓN ListeriaL
La listeriosis se define clínicamente cuando el microorganismo es aislado de sangre, líquido cefalorraquídeo (LCR) o de otros sitios del organismo humano o animal, que normalmente son estériles (placenta) (Low 1997). La susceptibilidad del hospedero juega un papel importante en la presentación de la enfermedad luego de la exposición a L. monocytogenes; de esta manera la mayoría de pacientes experimentan un defecto fisiológico ó patológico que afecta la inmunidad mediada por células T, lo que justifica la clasificación de L. monocytogenes como un patógeno oportunista. La fisiopatología de la infección por L. monocytogenes en humanos y animales no está totalmente esclarecida. La investigación de la fisiopatología de listeriosis requiere de un modelo animal en el cual el agente infeccioso pueda manifestar el mismo tropismo celular y tisular que en humanos; modelo en el que se observen los mismos efectos directos e indirectos que causan los daños inmunopatológicos (Lecuit 2002).
Existen dos estrategias para establecer un modelo para el estudio de la listeriosis: La primera es identificar una especie animal en la cual la interacción ligando-receptor sea específica y similar al humano. La segunda es generar un animal modificado genéticamente (transgénico), generalmente un ratón con un receptor no-funcional, en el cual el receptor humano pueda ser expresado bajo el control de un promotor tisular específico durante el proceso infeccioso (Lecuit 2002).
La inoculación intravenosa de L. monocytogenes en el ratón induce letalidad dosis-dependiente. Este modelo de infección ha sido usado por décadas para el estudio de la infección bacteriana intracelular y la inmunidad mediada por células (Mackaness 1969). En contraste, la inoculación oral es una vía ineficiente para inducir listeriosis sistémica, porque el paso de L. monocytogenes a través de la barrera intestinal es muy lento, confirmando que L. monocytogenes no es un microorganismo enteropatógeno para el ratón (Lecuit 2002).
Para investigar la interacción de proteínas de superficie de L. monocytogenes con la célula hospedero, se han probado diferentes modelos animales. Los modelos de ratón y rata han sido utilizados para el análisis de los factores de virulencia de L. monocytogenes que presentan homología con el humano, listeriolosina O (LLO), ActA, fosfolipasa C fosfatidilinositol (PI-PLC) y fosfolipasa C fosfatidilcolina (PC-PLC); pero han sido inapropiados para el estudio del mecanismo de interacción del receptor E-caderina en células epiteliales con la Internalina A de L. monocytogenes, ya que la naturaleza de uno de los aminoácidos que compone la E-caderina en ratón (ácido glutámico), es diferente al del humano (prolina), por esta razón estos modelos animales son resistentes a infección oral por L. monocytogenes (Lecuit 1999).
El segundo modelo utilizado para el estudio de la interacción de la internalina A en células epiteliales con la E-caderina de L. monocytogenes “in vivo”, fue el cultivo de células epiteliales de cobayo, donde se demostró que la entrada de L. monocytogenes a través de la internalina A es dependiente de si se expresa en la célula epitelial del hospedero la proteína E-caderina con una prolina en la posición 16; en este modelo animal, contrario a lo observado en ratón y rata en los cuales la E-caderina conserva el ácido glutámico en la posición 16, L. monocytogenes es capaz de inducir gastroenteritis, semejante a lo observado en humanos (Lecuit 1999).
En roedores se ha estudiado experimentalmente la tromboencefalitis por L. monocytogenes (Blanot et al., 1997). El descubrimiento de la especificidad por el hospedero y la elucidación del mecanismo de patogenia, ha conducido a la generación de ratones transgénicos que expresan receptores humanos que permiten la penetración de la bacteria a las células epiteliales, así como la posibilidad de atravesar los endotelios y alcanzar los órganos blancos de infección (Lecuit 2002).
Para evaluar el papel de la interacción internalina-E caderina en la habilidad de L. monocytogenes para cruzar la barrera intestinal, se diseñó un modelo de ratón transgénico (Lecuit 2001). El c-DNA de la E-caderina humana fue puesto bajo control del promotor del gen que codifica para la proteína de unión al ácido graso intestinal (iFABP), el cual es exclusivamente activo en enterocitos del intestino delgado post mitóticos, no proliferativos (Hermiston 1996). En este modelo transgénico L. monocytogenes interactuó directamente con la E-caderina enterocítica, se internalizó en las células, cruzó la barrera intestinal, se multiplicó en la lámina propia del intestino delgado y se diseminó a los nódulos linfáticos mesentéricos, hígado y bazo (Lecuit 2002).
La patogenicidad de L. monocytogenes se debe a la capacidad para adherirse, invadir y multiplicarse dentro de una gran variedad de células no fagocíticas (enterocitos, hepatocitos, fibroblastos, células endoteliales y células dendríticas). Esta propiedad, ha sido estudiada detalladamente en cultivos celulares, y es el punto de partida para el análisis fisiopatológico de la listeriosis humana (Gilot et al., 1999). La entrada de L. monocytogenes y la colonización de los tejidos del hospedero se lleva a cabo en cuatro etapas:
a. Cruce de la barrera intestinal: Antes de alcanzar el intestino, L. monocytogenes ingeridas tiene que soportar el ambiente adverso del estómago (Doyle 2001). Al menos 13 proteínas de estrés oxidativo y 14 proteínas de “shock” tóxico, son inducidas bajo condiciones de estrés en L. monocytogenes (Vázquez-Boland et al., 2001b).
L. monocytogenes posee un factor de transcripción sigma (sigma B) codificado por el gen sigB, homólogo a los encontrados en Staphylococcus aureus y Bacillus subtilis. Mutantes de este gen muestran un incremento de la sensibilidad al estrés ácido, sin embargo las cepas mutantes son capaces de diseminarse al hígado y al bazo de la misma manera que L. monocytogenes silvestre (Wiedmann et al., 1998).
Existe controversia con respecto al punto de entrada y el mecanismo de traslocación intestinal usado por L. monocytogenes. Rácz en 1972, infectó intragástricamente cobayos con 1010 UFC de L. monocytogenes, el análisis histológico reveló, que todos los animales desarrollaron enteritis. En etapas iniciales, la bacteria pudo detectarse principalmente en las células epiteliales absorbentes del área apical de las vellosidades, mientras en fases tardías, la bacteria se detectó en el interior de los macrófagos del estroma de las vellosidades, lo que sugirió que L. monocytogenes penetra el hospedero invadiendo el epitelio intestinal (Rácz et al., 1972).
En otros estudios usando ratones inoculados con 108 a 109 UFC, no se observó invasión del epitelio de las vellosidades intestinales, en cambio se evidenció colonización de las placas de Péyer (Marco et al., 1997), sugiriendo que L. monocytogenes usa las células M del epitelio como una entrada portal, al igual que se ha reportado para otros patógenos bacterianos (Siebers 1996). Otro estudio en 1998 demostró a través de un modelo murino y microscopía electrónica que L. monocytogenes penetra el hospedero por las células M dispuestas sobre las placas de Péyer (Jensen et al., 1998). Sin embargo, la penetración Listerial a este nivel no fue efectiva, porque únicamente un pequeño número de bacterias fueron observadas en asociación con el folículo asociado al epitelio sobre las placas de Péyer (Marco et al., 1992).
La traslocación intestinal de Listeria ocurre sin la formación de lesiones histológicas en el intestino del ratón (Marco et al., 1992). Esto sugiere que la fase intestinal que involucra la multiplicación bacteriana en la mucosa intestinal, no es requerida por L. monocytogenes para el desarrollo de infección sistémica.
Un estudio de infección intestinal en murino mostró que L. monocytogenes es traslocada a órganos internos en pocos minutos, demostrando que el cruce de la barrera intestinal ocurre en ausencia de replicación intraepitelial. El sitio preferencial para la replicación bacteriana fue las placas de Péyer. El foco de la infección se apreció a través de reacciones piogranulomatosas en el tejido folicular subepitelial y células mononucleares con bacterias visibles en su interior (Pron et al., 1998); los eventos de presentación antigénica tuvieron lugar en el intestino durante las fases tempranas de la colonización del hospedero; fenómeno que juega un papel importante en la resistencia adquirida a la reinfección posterior por exposición oral a L. monocytogenes (Low 1991).
b. Multiplicación en el hígado: Las células de Listeria cruzan la barrera intestinal a través de la linfa y la sangre hacia los nódulos linfáticos mesentéricos, el bazo y el hígado (Pron et al., 1998). Infecciones experimentales de ratones por vía intravenosa han mostrado que L. monocytogenes es retirada rápidamente de la corriente sanguínea por los macrófagos residentes en el bazo y el hígado (Cousens andWing 2000).
Más del 90% de bacterias son acumuladas en el hígado, capturadas por las células de Kupffer alineadas en las sinusoides. En experimentos de depleción del sistema inmune “in vivo”, los macrófagos residentes destruyen la mayoría de bacterias ingeridas, lo que resulta en la disminución del tamaño de la población bacteriana, durante las primeras 6 horas de infección (Ebe et al., 1999). Las células de Kupffer inician el desarrollo de la inmunidad anti-Listeria induciendo la proliferación dependiente de antígenos de linfocitos T y la secreción de citoquinas (Gregory 1990). No todas las células bacterianas son destruídas por macrófagos tisulares, y su número se incrementa en 2 a 5 días en órganos de ratón (De Chastellier 1994).
El sitio principal de multiplicación bacteriana en el hígado es el hepatocito (Conlan 1992). Mackannes (1962) fue el primero es demostrar que L. monocytogenes era capaz de sobrevivir y multiplicarse en macrófagos (Mackaness 1969). Existen dos vías posibles para el acceso de L. monocytogenes al parénquima del hígado después de la traslocación intestinal del microorganismo y el transporte por el torrente sanguíneo portal o arterial: Vía células de Kupffer, por la diseminación célula a célula o por la invasión directa de hepatocitos después de cruzar la barrera endotelial fenestrada que rodea los sinusoides hepáticos. Se ha demostrado “in vitro” que L. monocytogenes invade eficientemente los hepatocitos (Dramsi et al., 1995).
La microscopía electrónica de tejido hepático de ratones infectados sugiere que L. monocytogenes desarrolla completamente el ciclo infeccioso intracelular en los hepatocitos (Gaillard et al., 1996). El paso de hepatocito a hepatocito conduce a la formación de un foco infeccioso en el que L. monocytogenes se disemina a través del parénquima hepático sin entrar en contacto con los efectores humorales del sistema inmune; lo que pudiera explicar por qué los anticuerpos no son importantes en la inmunidad anti-Listeria (Edelson et al., 1999). Durante las etapas tempranas de colonización hepática, los polimorfonucleares son reclutados en los sitios de infección, en respuesta a la liberación de quimioatractantes por parte de los hepatocitos, formando microabscesos (Rogers et al., 1996). Los neutrófilos juegan un papel importante en el control de la fase aguda de la infección Listerial (Rogers 1993), y en la destrucción “in vivo” de Listeria en hepatocitos infectados (Conlan 1991).
Estudios en macrófagos murinos P388D1 revelan la activación de NF-KB; factor de transcripción que media la respuesta a la infección causada por L. monocytogenes a través de la regulación de genes involucrados en la respuesta inmune (Hauf et al., 1997). Las formas inactivas de NF-KB conocidas como IkBa y IkBb son fosforiladas por quinasas IkB, que inducen la traslocación de NF-KB dentro del núcleo de la célula hospedera, donde regula la expresión de citoquinas proinflamatorias (Hauf et al., 1994).
La activación de NF-kB en hepatocitos de ratón es esencial para eliminar a L. monocytogenes del hígado, este es un mecanismo de respuesta inmune innata local (Lavon et al., 2000). Entre dos y cuatro días después de la infección, los neutrófilos son reemplazados gradualmente por células mononucleares sanguíneas y linfocitos para formar los granulomas característicos que actúan como barreras físicas limitando el foco infeccioso e impidiendo la diseminación bacteriana de una célula a otra (Portnoy 1992).
Entre cinco a siete días de postinfección, L. monocytogenes comienza a desaparecer en los órganos internos de ratón como resultado de la activación de macrófagos por medio del interferón gamma (IFN-g) y la inducción de la respuesta inmune adquirida mediada por linfocitos T CD8+ (Gregory 2000). Los linfocitos T citotóxicos reaccionan contra los epítopes de los antígenos del complejo mayor de histocompatibilidad clase I o II presentes en la membrana de la célula blanco, desencadenando la apoptosis de los hepatocitos infectados (Parham 1997).
Wagner y Czuprynski (1993) observaron la expresión temporal de citoquinas en el hígado de ratones infectados con L. monocytogenes; IFN-g, el factor de necrosis tumoral (TNF-a) y la interleuquina 10 (IL-10), fueron inducidos un día después de la infección, las IL-2 y la IL-4 producidas por los LT fueron expresadas el primer día, pero suprimidas tres días después de la infección.
La protección contra L. monocytogenes también involucra los linfocitos T CD4+ TH1 (Geginat et al., 1998) y mecanismos innatos como la activación de células T asesinas (“natural killer”). El IFN-g y la IL-12 producidos por los macrófagos inducen la formación de LT CD4+ TH1 (Emoto et al., 1997).
Las lipoproteínas bacterianas pueden ser potentemente proinflamatorias e inducen respuestas inmunitarias innatas y adaptativas en mamíferos. Dentro de las lipoproteínas descritas para L. monocytogenes está la TcsA que es presentada por el complejo mayor de histocompatibilidad tipo II (CMH II) y media la activación de linfocitos T CD4 (Aliprantis 1999).
La exposición a especies no patogénicas como L. innocua aumenta la respuesta mediada por células T contra L. monocytogenes ya que ambas especies comparten el epítope p60 que es reconocido por los linfocitos T citotóxicos (Geginat et al., 1999). Si la infección no es controlada por una respuesta inmune adecuada en el hígado, como puede ocurrir en individuos inmunosuprimidos, la proliferación ilimitada de L. monocytogenes en el parénquima hepático puede resultar en la liberación de bacterias dentro de la circulación (Doyle 2001; Vázquez-Boland et al., 2001a; Vázquez-Boland et al., 2001b).
L. monocytogenes es un patógeno multisistémico que puede infectar un amplio rango de tejidos hospederos. Sin embargo, las principales formas clínicas de listeriosis muestran que este microorganismo posee tropismo por el útero grávido y el sistema nervioso central (Vázquez-Boland et al., 2001b).
c. Colonización de útero grávido y feto: El aborto y la muerte del neonato debido a Listeria spp., han sido reproducidos experimentalmente por inoculación intravenosa, oral y respiratoria de Listeria en animales gestantes susceptibles, como ovejas, reses, conejos, cobayos, ratones y ratas, de esta manera se demuestró que L. monocytogenes accede al feto por penetración hematógena de la barrera placentaria (Abram 1997). En ratones grávidos, la bacteria primero invade la membrana basal y progresa hacia el vello placental, donde causa infiltración inflamatoria y necrosis (Abram 1997). Los macrófagos no actúan como efectores en la placenta murina, mientras los neutrófilos son la población celular más importante contra Listeria (Guleria 2000).
En humanos la infección placentaria se caracterizada por numerosos microabscesos e inflamación de las vellosidades con necrosis focal (Parkassh et al., 1998). La colonización de la membrana trofoblástica seguida por la traslocación a través de la barrera endotelial, permite a la bacteria alcanzar la corriente sanguínea fetal, conduciendo a una infección generalizada y la posterior muerte del feto en útero o la muerte prematura del neonato infectado con lesiones piogranulomatosas miliares (Vázquez-Boland et al., 2001b).
La depresión local de la respuesta inmune celular en la placenta, puede contribuir al aumento de la susceptibilidad a la infección uterina por L. monocytogenes (Vázquez-Boland et al., 2001b). En roedores, la gestación reduce la resistencia a L. monocytogenes y prolonga el curso de la infección primaria en el hígado (Abram 1997); lo anterior se correlaciona con la deficiencia en la producción de IFN-g que resulta en la invasión Listerial de la placenta y los tejidos fetales (Nakane et al., 1985).
d. Invasión del cerebro: en humanos, la infección del sistema nervioso central se presenta en forma de meningitis, Sin embargo, esta meningitis está asociada frecuentemente con la presencia de focos infecciosos en el parénquima cerebral, especialmente en el tallo cerebral lo que sugiere que L. monocytogenes tiene tropismo por el tejido nervioso (Lorber 1996).
Aunque se presenta infiltración mononuclear y linfocítica de las meninges, esta condición ocurre como una extensión del proceso cerebral, y las lesiones macroscópicas pueden restringirse a áreas basales, al cerebro blando y al cerebelo. En humanos, al igual que en rumiantes se desarrolla una cerebritis que involucra el romboencéfalo (Lorber 1996). El neurotropismo y la predilección de L. monocytogenes por el romboencéfalo se ha demostrado en los rumiantes, en los cuales la infección en el sistema nervioso central, en contraste a la situción en humanos, se desarrolla como encefalitis. En estos animales, el foco infeccioso es restringido a la médula oblongata y el cordón espinal (Vázquez-Boland et al., 2001b).
L. monocytogenes invade el cerebro por migración centrípeta a lo largo de los nervios craneales. La parálisis unilateral del nervio craneal es característica de romboencefalitis en rumiantes, conduciendo al desarrollo de la enfermedad “circling” que se caracteriza por tortículis involuntaria y andar sin rumbo, en círculos como resultado de las lesiones del tallo cerebral (Wesley 1999).
Las lesiones cerebrales en la meningoencefalitis Listerial son típicas y muy similares en humanos o animales y consisten en nódulos perivasculares de infiltrados inflamatorios, compuestos de células mononucleares, neutrófilos dispersos y linfocitos; microabscesos parenquimales y focos de necrosis. Las bacterias están generalmente ausentes en las áreas perivasculares de inflamación y son abundantes en el parénquima cerebral alrededor de las áreas necróticas (Jungi et al., 1997). La invasión por L. monocytogenes de neuronas cultivadas “in vitro” es un evento relativamente raro (Dramsi andCossart 1998).
L. monocytogenes es capaz de invadir células del endotelio microvascular cerebral de humanos “in vitro” e inducir la activación de moléculas para la adhesión endotelial (E-selectina, ICAM-1 y VCAM-1) que conducen al incremento de la unión de polimorfonucleares neutrófilos a las células endoteliales infectadas (Kayal et al., 1999; Greiffenberg et al., 2000). Experimentos de depleción en ratones usando un anticuerpo monoclonal específico, mostró que los neutrófilos juegan un papel crítico en la eliminación de L. monocytogenes de los focos infecciosos en el cerebro (López et al., 2000).
POBLACIÓN DE RIESGO
L. monocytogenes posee predilección por personas con inmunidad celular disminuida, las poblaciones de riesgo son: mujeres en estado de gravidez (Bartfield 2000; DiMaio 2000), ancianos, neonatos (Becroft 1971); personas inmunocomprometidas (cáncer, transplantes renales, SIDA, diabetes, terapias inmunosupresoras, alcoholismo). (Graham et al., 2002; Morritt et al., 2002; Nolla-Salas et al., 2002; Vander et al., 2002).
La listeriosis en adultos excepto mujeres embarazadas, es asociada en muchos casos (>75%) con: neoplasias (leucemia, linfoma o sarcoma) y quimioterapia antineoplástica, terapia inmunosupresora (transplante de órganos o uso de corticoides) (Patel 1997), enfermedad hepática crónica (cirrosis o alcoholismo) (Kendall et al., 1972), endocarditis, enfermedad del riñón, diabetes y enfermedad del colágeno (lupus) (Rocourt 1996; Spyrou et al., 1997).
La infección por el virus de inmunodeficiencia humana (VIH) es también un factor de riesgo para listeriosis. El SIDA es una condición de predisposición que oscila entre el 5 y el 20% de los casos de listeriosis en adultos excepto mujeres embarazadas. El riesgo de contraer listeriosis es de 300 a 1000 veces mayor para pacientes con VIH que para la población general. Aunque muchos de los casos son asociados con factores de riesgo, hay algunos pacientes adultos donde los factores de predisposición no han sido determinados (Goulet et al., 1993).
CARACTERÍSTICAS CLÍNICAS DE LISTERIOSIS HUMANA
Dos formas básicas de presentación de listeriosis pueden distinguirse: listeriosis perinatal y listeriosis en el paciente adulto. En ambas instancias, las formas clínicas predominantes corresponden a infección diseminada o infección local en el sistema nervioso central (SNC). Los alimentos contaminados son la mayor fuente de infección tanto en casos esporádicos como epidémicos (Rocourt 1996). La dosis mínima requerida de Listeria monocytogenes para causar infección clínica en humanos no ha sido determinada, sin embargo el gran número de L. monocytogenes detectadas en los alimentos responsabilizados de casos esporádicos y epidémicos de listeriosis (106) sugiere que es alta (Vázquez-Boland et al., 2001b).
Niveles de 102 a 104 células de L. monocytogenes por gramo de alimento han sido asociados con listeriosis en humanos, especialmente en pacientes inmunosuprimidos, ancianos y mujeres embarazadas (McLauchlin et al., 1991; Pinner et al., 1992). Sin embargo, la dosis infectiva puede variar dependiendo de la patogenicidad y virulencia de la cepa involucrada y los factores de riesgo y susceptibilidad del hospedero (Vázquez-Boland et al., 2001b). La dosis infectiva de L. monocytogenes depende de factores como el estado inmunológico del paciente, la concentración de patógeno en el alimento, la virulencia de la cepa, y la cantidad de alimento consumido (McLauchin 1996).
El curso clínico de la infección usualmente comienza alrededor de 20 horas después de la ingestión del alimento contaminado (Dalton et al., 1997). El período de incubación para la enfermedad invasiva es generalmente entre 20 y 30 días (Riedo et al., 1994). Períodos de incubación similares han sido reportados en animales tanto para la enfermedad gastroentérica como para la invasiva (Vázquez-Boland et al., 1996). Los síntomas gastrointestinales como naúseas, vómito y diarrea pueden preceder a formas más serias de listeriosis o pueden aparecer de 9 a 48 horas luego de la infección. El período de incubación en personas adultas es de 3 a 70 días, en bebés infectados al momento de nacer el período de incubación es de 1 a 4 semanas después del nacimiento (Graham et al., 2002).
La listeriosis fetomaternal y listeriosis neonatal resultan de la transmisión de la infección madre-feto por vía transplacentaria y desarrollo de corioamnionitis. La mayoría de los casos de listeriosis en el feto ocurren después del quinto mes de embarazo, aunque se han presentado casos de infecciones tempranas que ocasionan daños en el embrión (Silver 1998). La listeriosis materna puede precipitar el trabajo de parto y provocar un óbito fetal o un parto pretérmino de un feto infectado. La mujer puede portar el microorganismo en el tracto genital por algún tiempo, ocasionando complicaciones en embarazos posteriores (Schuchat 1997).
La infección neonatal por Listeria monocytogenes se puede manifestar en dos formas:
a) Listeriosis de inicio temprano: El período de incubación es de 1.5 días y la infección ocurre in útero; se asocia con aborto espontáneo, nacimiento prematuro, bajo peso al nacer, bronconeumonía o septicemia al nacimiento, o muerte del recién nacido por una infección diseminada conocida como granulomatosis infantiséptica, caracterizada por la presencia de microabscesos piogranulomatosos diseminados particularmente en hígado, bazo y vellosidades coriónicas; con mortalidad del 35 al 10%. Los síntomas generalmente incluyen dolor pulmonar, debilitamiento del corazón, deficiencias respiratorias, cianosis, vómito, convulsiones y descarga temprana del meconio (Schlech 1996). Las madres de estos productos con frecuencia tienen antecedente de infección tipo influenza con fiebre o infección de vías urinarias en las semanas que preceden el parto. Durante esta etapa los cultivos de sangre materna son positivos para Listeria monocytogenes (Klatt et al., 1986).
b) En menor frecuencia se observa la listeriosis neonatal tardía (10 al 15% de casos perinatales), generalmente ocurre de 1 a 8 semanas post-parto e involucra síndrome febril acompañado por meningitis y en algunos casos gastroenteritis y neumonía, presumiblemente como resultado de la aspiración de exudados maternales contaminados durante el parto, aunque se han reportado casos en donde la enfermedad es adquirida mediante transmisión horizontal por fomites o por personal médico en unidades neonatales, con mortalidad del 13 al 43% en pacientes tratados y de 100% en pacientes no tratados (Slutsker 1999).
La mortalidad en casos de listeriosis neonatal es baja (10-20%), pero puede dejar secuelas como hidrocefalia o retardo psicomotor (Evans et al., 1984). L. monocytogenes es una de las tres causas principales de meningitis bacterial en neonatos (Lorber 1996); en adultos, afecta el sistema nervioso central en un 55 a 70% de los casos, normalmente se desarrolla como una meningoencefalitis acompañada por trastornos severos de la conciencia, desórdenes en el movimiento y en algunos casos parálisis en los nervios craneales (Armstrong 1993). Cuando L. monocytogenes es diseminada vía sanguínea al sistema nervioso central, puede ocasionar meningitis supurativa aguda, cerebritis focal o multifocal, meningoencefalitis, absceso cerebral o espinal y listeriosis bulbar o romboencefalitis. La listeriosis es comúnmente caracterizada por formación de listeromas y necrosis focal. El tamaño, número y sitio de las lesiones varía de persona a persona y está relacionado con el modo de infección, la dosis del microorganismo, la edad y la resistencia del paciente (Mrowka et al., 2002).
La forma encefalítica en la cual el microorganismo es aislado con dificultad del fluido crebroespinal, es común en animales pero rara en humanos. El curso de la enfermedad es usualmente bifásico, con una fase subfebril inicial de 3 a 10 días en las cuales se presentan dolores de cabeza, desórdenes visuales, malestar general; seguida por una segunda fase de síntomas severos de romboencefalitis (Vázquez-Boland et al., 2001b).
El porcentaje de mortalidad por infecciones en el sistema nervioso central es alrededor del 20% pero pueden llegar a elevarse hasta un 40-60% si está asociado con enfermedades recurrentes que debilitan el sistema inmune. Se ha estimado que el 10% de la población que adquiere meningitis bacterial es causada L. monocytogenes. Debido a la efectiva vacunación contra Haemophylus influenzae; L.monocytogenes es ahora la causa mas común de infección meningeal en adultos después de Streptoccocus pneumoniae, Neisseria meningitidis y el Streptococo del grupo B. Sin embargo, en ciertos grupos de alto riesgo, como pacientes con cáncer, L. monocytogenes es la principal causa de meningitis bacterial (Schuchat 1997).
Otra forma clínica frecuente de listeriosis en algunos pacientes, es la bacteriemia o septicemia (15 del 50% de los casos), con un alto porcentaje de mortalidad hasta de 70%, si esta asociado con enfermedades recurrentes que debilitan el sistema inmune. Existen otros formas clínicas atípicas en el 5 al 10% de los casos, como la endocarditis (tercera forma más frecuente), miocarditis, arteritis, neumonía, pleuritis, hepatitis, colecistitis, peritonitis, abscesos localizados, artritis, osteomielitis, sinusitis, otitis y conjuntivitis (Low 1997; Slutsker 1999).
Se ha identificado una forma primaria de infección cutánea por L.monocytogenes la cual es caracterizada por una erupción piogranulamotosa; esta forma ocurre esporádicamente entre granjeros y veterinarios y se adquiere por contacto directo con el tracto genital o la placenta de vacas que han abortado debido a infección por Listeria (Marth 1988; Ireton et al., 1996).
SENSIBILIDAD A ANTIBIÓTICOS
El patrón de sensibilidad a los antibióticos de L. monocytogenes ha permanecido relativamente estable con el paso de los años (Cherubin et al., 1991). Generalmente, este microorganismo es sensible “in vitro” a una amplia gama de antibióticos como penicilina, ampicilina, gentamicina, eritromicina, tetraciclinas, rifampicina, cotrimoxazol y vancomicina. Las fluorquinolonas y las cefalosporinas actuales presentan una pobre actividad, especialmente las de tercera y cuarta generación como cefotaxima y cefepima; todas las cepas de Listeria monocytogenes son resistentes a fosfomicina (Kalstone 1991).
Se han descrito resistencias a macrólidos y a tetraciclinas en algunos aislamientos; suelen estar codificadas por plásmidos, aunque también hay casos de resistencia a tetraciclinas codificada cromosómicamente. La rifampicina posee buena actividad “in vitro” pero selecciona cepas resistentes durante el tratamiento con mucha facilidad. Aunque se han descrito fracasos clínicos en tratamientos con penicilina o ampicilina, no se ha encontrado ninguna cepa resistente a estos antibióticos (Kalstone 1991).
La mayor parte de los antibióticos se comportan como bacteriostáticos frente a L. monocytogenes. En particular los ß-lactámicos tienen un gran intervalo entre la concentración mínima inhibitoria (CMI) y la concentración mínima bactericida (CMB). Sólo se ha observado actividad bactericida en los aminoglucósidos, glucopéptidos y cotrimoxazol (Hofer 1988).
TRATAMIENTO CONTRA L. monytogenes
Al ser la listeriosis una enfermedad relativamente rara en humanos, no hay estudios prospectivos y controlados que establezcan el mejor tratamiento antibiótico. Se ha visto que Listeria monocytogenes puede ser refractaria a los mecanismos bactericidas de muchos antibióticos porque es intracelular y usa este mecanismo para multiplicarse y protegerse de los antibióticos que se encuentran en el fluido extracelular; sólo pocos agentes pueden penetrar, acumularse y alcanzar el citosol de las células que hospedan este microorganismo (Charpentier 1997).
Actualmente se considera que los antibióticos de primera elección son la penicilina o la ampicilina (â-lactámicos), solas o asociadas a gentamicina (aminoglucósido). Se han descrito fallos terapéuticos con estos antibióticos, pero nunca se ha demostrado resistencia al compuesto ß-lactámico utilizado; el antibiótico de segunda elección es eritromicina (macrólido) (Hof et al., 1997).
En las enfermedades graves como la cerebritis o la granulomatosis infantiséptica, el inicio oportuno del tratamiento es fundamental para el control de la infección. Estudios “in vitro” han demostrado sinergia entre ampicilina y penicilina con aminoglucósidos (Moellering et al., 1972). Esta asociación debe utilizarse en casos de granulomatosis infantiséptica, sepsis neonatal, meningoencefalitis y en pacientes inmunosuprimidos (Carvajal et al., 1989).
La combinación de trimetoprim y sulfametoxazol se ha utilizado con éxito en pacientes alérgicos a penicilinas, considerándose en la actualidad la terapia alternativa en esta circunstancia. Hay estudios donde se relaciona la existencia de un plásmido de 3.7Kb en cepas de Staphylococcus haemolyticus (colonizador de la piel), que codifica para la dihidrofolato reductasa y que confiere resistencia a sulfametoxazol. También se ha observado que el aumento del uso de sulfametoxazol en profilaxis para toxoplasmosis en pacientes transplantados o VIH positivos, a pesar de que puede disminuir el número de casos de listeriosis en adultos, también pudede generar resistencia (Charpentier 1997).
La duración del tratamiento no está clara. Tras dos semanas de terapia se han descrito recurrencias en pacientes inmunosuprimidos; por lo que parece conveniente prolongar la terapia entre tres y seis meses. En general dos semanas parecen ser suficientes en bacteremias mientras que en meningitis se deberían utilizar ciclos más largos (Hof et al., 1997).
Considerando que la L. monocytogenes comparte mecanismos de parasitismo intracelular con otros organismos, su caracterización puede ser de utilidad para hallar nuevos agentes terapéuticos. Vázquez Boland (2001b) ha identificado el primer factor de virulencia bacteriano que está implicado específicamente en la fase de proliferación intracelular y que es compartido por otros microorganismos. De esta forma, si se desarrollan inhibidores específicos de este mecanismo, se podría frenar la proliferación “in vivo” de ésta y de otras bacterias patógenas.
ORGANIZACIÓN GENÉTICA Y EVOLUCIÓN DE LOS DETERMINANTES DE VIRULENCIA DE L. monocytogenes
Aunque se han reportado plásmidos en Listeria spp. (Poyart-Salmeron et al., 1990; Lebrun et al., 1994); todos los determinantes de virulencia identificados hasta el momento están codificados cromosómicamente. Los genes de virulencia de Listeria spp., se organizan dentro de unidades genéticas conocidas como islas de patogenicidad (PAIs). Las PAIs son adquiridas por la bacteria por mecanismos de transferencia de información genética horizontal, algunas veces como parte de un elemento móvil genético, por lo cual son importantes en la evolución de la virulencia bacteriana (Hacker 2000).
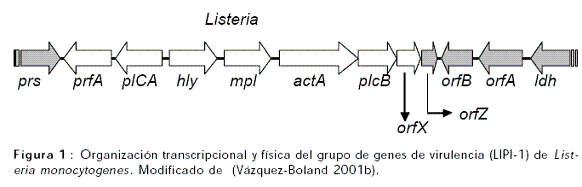
Una serie de factores de virulencia son producidos por L. monocytogenes para facilitar el proceso invasivo (Portnoy et al., 1992). Seis de los factores de virulencia responsables del parasitismo intracelular de L. monocytogenes (prfA, plcA, hly, mpl, actA y plcB) están organizados en una isla cromosomal de 9kb conocida como grupo de genes de virulencia PrfA dependiente; isla denominada como isla 1 de patogenicidad de Listeria (LIPI-1) (Figura 1) (Vázquez-Boland et al., 2001a; Vázquez-Boland et al., 2001b).
El locus de virulencia está formado en tres unidades transcripcionales (Figura 1). La posición central está ocupada por el monocistrón hly, que codifica para una citolisina sulfidrilo-activada (listeriolisina O) requerida para la ruptura de la vacuola fagocítica y la liberación de la bacteria dentro del citoplasma; prerequisito para la proliferación intracelular de L. monocytogenes (Cossart 2001c; Vázquez-Boland et al., 2001a). Corriente abajo del monocistrón hly, y en el mismo sentido de transcripción se encuentra el operón lecitinasa de 5.7Kb que comprende tres genes: mpl, actA y plcB y tres pequeños marcos de lectura abiertos (ORFs) adicionales (Figura 1) (Vázquez-Boland et al., 1992a; Vázquez-Boland et al., 1992b). El gen ActA codifica para la proteína ActA, el factor responsable de la motilidad basado en actina y la diseminación célula a célula de L. monocytogenes (Lasa et al., 1998). El gen plcB codifica para la fosfolipasa C fosfatidilcolina específica (PC-PLC), que media la disolución de la doble membrana de los fagosomas secundarios, formados después de la diseminación célula a célula. El gen mpl codifica para la proteasa mpl, la cual procesa extracelularmente el propéptido inactivo de la PC-PLC (Smith et al., 1995).
Los tres productos del operón (mpl, ActA y plcB), están involucrados en una función que es esencial en la patogénesis de L. monocytogenes: La diseminación de la bacteria de una célula a otra. Esta función le permite a la bacteria evadir el compartimiento extracelular y por tanto, los efectores humorales del sistema inmune durante su diseminación en tejidos del hospedero (Portnoy 1992).
Corriente arriba del monocistrón hly, y transcrito en dirección opuesta, está el operón plcA-prfA (Figura 1). El primer gen de este bicistrón (plcA), codifica para la fosfolipasa C fosfatidilinositol específica (PI-PLC), la cual media la desestabilización de los fagosomas primarios junto con la LLO y la PC-PLC (Smith et al., 1995). El gen prfA codifica para la proteína PrfA, factor de transcripción estructural y funcionalmente relacionado a la proteína Crp (CAP) de Enterobacterias. El gen prfA de 27kDa es el activador transcripcional de los genes de virulencia de L. monocytogenes (Figura 1) (Goebel 2000a). La expresión del regulón de virulencia vía PrfA depende de diversas señales ambientales. Estas señales de activación incluyen temperatura alta (37ºC) (Leimeister-Wachter et al., 1992), condiciones de estrés (Sokolovic et al., 1990), reclusión de componentes del medio de crecimiento celular por carbón activado (Ripio et al., 1996), contacto con células del hospedero y el ambiente citoplasmático eucariótico (Renzoni et al., 1999).
El modelo actual de expresión del regulón de virulencia vía PrfA de L. monocytogenes predice un mecanismo que involucra la activación alostérica de PrfA por un cofactor putativo de bajo peso molecular, cuyos niveles dependen de las condiciones ambientales (Vega et al., 1998). La activación del PrfA conduce a la síntesis de más proteína PrfA por retroalimentación positiva, mediada por el promotor dependiente de PrfA, el cual gobierna la síntesis del RNAm del bicistrón plcA-prfA (Vega et al., 1998).
VÍAS EVOLUTIVAS DE LOS GENES DE VIRULENCIA
Análisis filogenéticos basados en el RNA ribosomal 16S (RNAr 16S) y 23S (RNAr 23S) han revelado que el género Listeria comprende dos grupos evolutivos, uno relativamente distante y que corresponde a L. grayi y el otro que incluye por un lado L. monocytogenes (patógena) y L. innocua (no patógena) y por el otro L. ivanovii (patógena), L. seeligeri y L. welshimeri (no patógenas) (Figura 2) (Vázquez-Boland et al., 2001b).
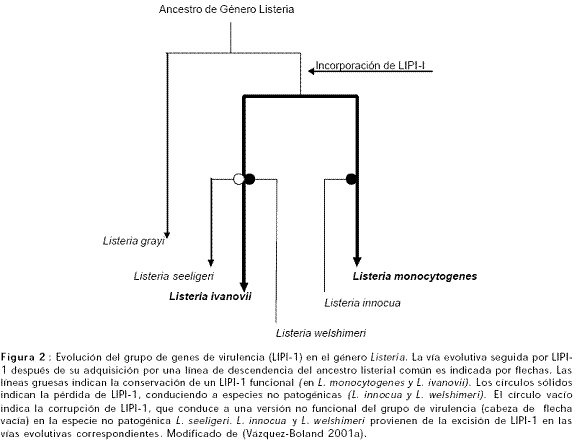
L. ivanovii desarrolla un ciclo de vida intracelular similar a L. monocytogenes ya que lleva una copia de LIPI-1. Las estructuras genéticas de LIPI-1 de L. monocytogenes y L. ivanovii son idénticas, pero las secuencias de ADN exhiben únicamente un 73 a un 78% de similaridad, un grado de divergencia compatible con la distancia genética que separa ambas especies (Figura 2) (Vázquez-Boland et al., 2001b).
El LIPI-1 no está presente en el genoma de las especies no patogénicas del género Listeria, a excepción de L. seeligeri en la cual hay una disrupción de la autoregulación positiva en el proceso de activación del LIPI-1 (Chakraborty et al., 2000; Vázquez-Boland et al., 2001a).
El resultado de LIPI-1 en cada una de las dos líneas de descendencia es importante en la diversificación posterior del género; estabilización en L. monocytogenes y L. ivanovii; deleción temprana en L. innocua y L. welshimeri e inactivación funcional en L. seeligeri (Vázquez-Boland et al., 2001b).
CICLO INFECCIOSO INTRACELULAR DE L. monocytogenes
La razón por la que L. monocytogenes causa infección está explicada en la capacidad para inducir fagocitosis en células del sistema mononuclear fagocítico, seguida de la replicación dentro de estas y la transferencia directa a células vecinas (Drevets 1999). El ciclo de infección se divide en cuatro etapas: Internalización, Evasión de la vacuola intracelular, Nucleación de filamentos de actina y Expansión de célula a célula (Vázquez-Boland et al., 2001b).
A. Internalización: El ciclo comienza con la adhesión a la superficie de la célula eucariota y la subsecuente penetración de la bacteria dentro de la célula hospedero, L. monocytogenes es tomada por las células del hospedero a través de fagocitosis (Finlay 1997).
Las bacterias invasivas han sido clasificadas dentro de dos grupos de acuerdo al cambio morfológico que ocurre en el sitio de entrada:
o Mecanismo “trigger” (disparador): Una bacteria en contacto con una célula, libera factores de virulencia directamente dentro del citoplasma del hospedero; factores que activan la maquinaria del citoesqueleto y vías de transduccción de señales que forman una membrana rugosa que internaliza la bacteria en un tipo de macropinocitosis.
o Mecanismo “zipper” (cremallera): Un ligando de la bacteria interactúa con una molécula de superficie sobre la célula hospedero, generalmente una proteína involucrada en la adhesión celular y/o activación de la maquinaria del citoesqueleto. La interacción de este receptor con el ligando de la bacteria induce rearreglos locales en la actina del citoesqueleto y otras señales que culminan en el desarrollo del cuerpo bacterial por la membrana plasmática. Listeria pertenece a la segunda categoría de bacterias invasivas (Machesky 2001).
El único mecanismo conocido que permite la unión covalente de las proteínas de superficie de la pared celular de bacterias Gram-positivas a la célula hospedero requiere de una secuencia motivo conservada LPXTG (Leu-Pro-X-Thr-Gly, donde X es cualquier aminoácido) seguida de un dominio hidrofóbico de 20 aminoácidos y una cola de aminoácidos cargados positivamente (55aa). Esta secuencia retiene el polipéptido en la membrana celular. Dicha retención es seguida por el anclaje entre la treonina y los residuos de glicina del motivo LPXTG y la unión directa del grupo carboxilo de los residuos de treonina al ácido diaminopimélico encontrado en la pared celular bacteriana. Esta reacción es catalizada por una proteína de membrana conocida como sortasa (Cossart 2000b; Dhar 2000; Cabanes et al., 2002).
En el genoma de L. monocytogenes se han detectado 41 genes que codifican para las proteínas LPXTG. La primera proteína LPXTG identificada en este microorganismo fue la InlA, la cual media la internalización bacterial a través de las células epiteliales (Gaillard 1991).
La internalina A y la proteína de adhesión a Listeria (LAP) de 104KDa son proteínas de superficie de Listeria requeridas para la penetración al interior de las células no fagocíticas (Nivia et al., 1999; Pandiripally et al., 1999; Santiago et al., 1999). La internalina A forma parte del la familia multigénica de internalinas junto con las internalinas E, F, G y H las cuales no están involucradas en el proceso invasivo de L. monocytogenes pero son importantes para la colonización del tejido del hospedero “in vivo” (Kajava 1998; Raffelsbauer 1998; Schubert 2001).
La internalina A, proteína de superficie de 800 aminoácidos, está unida covalentemente al peptidoglicano y contiene un dominio amino terminal repetitivo rico en leucina (LRR) seguido de una región inter-repetitiva conservada (IR), el carbono terminal comprende dos y medio repeticiones de 75 aminoácidos, seguida de una región que contiene un motivo LPXTG que permite la unión covalente de la proteína al péptidoglicano (Cossart et al., 2003).
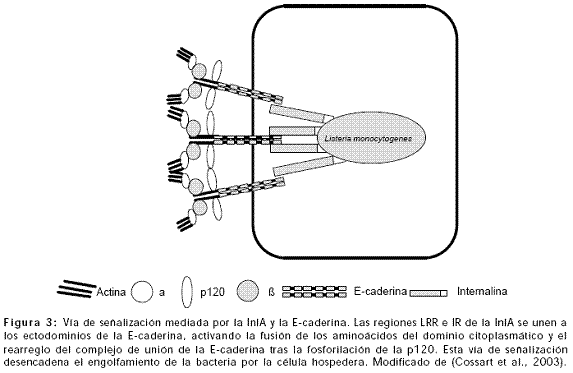
El receptor en la célula epitelial, para la internalina A es la proteína transmembranal E-caderina, glicoproteína transmembranal que contiene cinco dominios extracelulares de caderina (Ectodominios, EC1-EC5) y un dominio intracitoplasmático que regula la adhesión célula a célula y es dependiente de calcio. La E-caderina forma un dímero permitiendo las interacciones homofílicas mediadas por los dominios EC1 y EC2. Diferentes proteínas interactúan con el dominio citoplasmático formando un complejo de unión que se activa en forma de cascada, p120 è Vav2, Shcèb catenina, b cateninaèa catenina, a cateninaèafadina, b cateninaèIQGAP. La interacción del IQGAP, efector de las GTPasas Rac-1 y Cdc42 con la b catenina regula la conexión de la E-caderina con el citoesqueleto de actina, inhibiendo la asociación de la catenina con la a catenina. Adicionalmente, proteínas como la a-actinina, zyxina, vinculina, VASP/MENA, ZO-1, complejo Arp2/3 y algunas proteínas transmembranales adicionadas a la nectina completan este complejo (Kussel-Andermann 2000; Vasioukhin 2000; Nagafuchi 2001; Vasioukhin 2001).
Las regiones LRR e IR de la InlA se unen a los ectodominios de la E-caderina, activando la fusión de los aminoácidos del dominio citoplasmático y el rearreglo del complejo de unión de la E-caderina tras la fosforilación de la p120. Esta vía de señalización desencadena la entrada de la bacteria a la célula hospedero. Adicionalmente se estimulan proteínas transmembranales como la vezatina y sus ligandos, la miosina VIIA y componentes de la matriz extracelular como el heparán sulfato que estimulan la fagocitosis de L. monocytogenes y que sugieren que el proceso de internalización requiere de un motor de miosina (Figura 3) (Kussel-Andermann 2000; Lecuit 2000; Cossart et al., 2003).
Dos vías de señalización sinérgicas son activadas por Listeria cuando interactúa con una célula de mamífero, la primera mediada por la InlA y la otra por otra internalina denominada InlB, la cual es una proteína de 630 aminoácidos que contiene un péptido señal, una región LRR constituída por una región cap N-terminal pequeña y una región tubular extensa ligeramente curva constituída de motivos lámina b seguidos de hélices, una región IR que simula una inmunoglobulina y una región carboxi-terminal formada por tres módulos altamente conservados de 80 aminoácidos que incluyen un dipéptido inicial Gly-Trp (módulo GW), y que están unidos de manera no covalente a los ácidos lipoteicoicos de la pared celular bacteriana (Jonquieres 1999; Schubert 2001; Bierne 2002; Cabanes et al., 2002; Marino 2002). El módulo GW de la InlB media la unión específica al gC1qR el cual puede moverse alternativamente entre la mitocondria, superficie celular y el núcleo (Van Leeuwen 2001; Marino 2002).
Met (receptor para el factor de crecimiento hepatocito (HGF)) es un heterodímero compuesto de una subunidad a de 45KDa y una subunidad de 145KDa. La subunidad a y la región amino terminal de la subunidad b forman el dominio extracelular. El sitio de autofosforilación necesario para la funcionabilidad del receptor es la subunidad b que tiene un dominio transmembranal y una cola citoplasmática con un dominio rico en tirosina y con actividad tirosin-quinasa. La InlB interactúa con el dominio extracelular del receptor Met a través del dominio LRR (Figura 4) (Cossart 2001a; Cossart et al., 2003).
Recientemente los glicosaminoglicanos (GAGs) han sido identificados como un tercer tipo de ligando para la InlB. Estos compuestos sirven para “decorar” los proteoglicanos sobre la superficie de las células de mamíferos e incrementar la eficiencia de la entrada bacteriana. Los GAGs promueven la oligomerización y el almacenamiento en la superficie celular de factores de crecimiento tales como el HGF; adicionalmente protegen al HGF de proteasas extracelulares. La InlB se une a los GAGs a través de los módulos GW. La presencia de GAGs incrementa la activación del receptor Met dependiente de InlB (Figura 4) (Jonquieres 2001).
La InlB permanece unida de manera no covalente a los ácidos lipoteicoicos de la pared celular bacteriana, donde es protegida de la degradación proteolítica y de agentes externos agresivos. En proximidad a la célula hospedero, la InlB puede disociarse de la superficie bacteriana por interacción con los GAGs. El dominio LRR soluble de la InlB interactúa con el dominio extracelular del receptor Met y el módulo GW soluble de la InlB interactúa con el receptor gC1qR induciendo la fosforilación de tirosinas de las proteínas Gab1 y Cbl y Shc y la formación de complejos con estas proteínas fosforiladas y la subunidad p85 del PI 3-quinasa que median las señales necesarias para la reorganización del citoesqueleto y otros eventos involucrados en la entrada a la célula hospedero. Los eventos que inducen la reorganización del citoesqueleto involucran las cascadas Rac=>Wave=>complejo Arp2/3; Rac-1=>PAK=>SSH=>factor ADF (cofilin) que estimulan el ensamblaje de filamentos de actina, el desarrollo del cuerpo bacterial por la membrana plasmática y el proceso de internalización (Figura 4). Simultáneamente se activan otras enzimas como la fosfolipasa C-g1 y el factor NF-kB que pueden afectar el destino de la bacteria en la célula y/o el comportamiento de la célula hospedero (Bierne 2001; Jonquieres 2001; Mansell 2001; Eden 2002; Cossart et al., 2003).
B. Evasión de la vacuola intracelular: Una vez que L. monocytogenes ha sido fagocitada por un macrófago, en pocas horas se cubre de filamentos de actina. La cubierta de actina se organiza para formar apéndices de actina F, facilitando el desplazamiento de L. monocytogenes en el interior del macrófago y la posterior diseminación a macrófagos contiguos. Las condiciones que existen en el fagolisosoma, pH de 5.5+/-0.2 y bajas concentraciones de hierro no le permiten multiplicarse, pero inducen la secreción de Listeriolisina O (LLO) (Dramsi 2002).
La Listerilosina O es una citolisina sulfidrilo-activada de 58kDa, codificada por el gen hly, oxígeno lábil, formada por 27 aminoácidos y cuya secuencia es similar a las secuencias PEST frecuentemente encontradas en proteínas de humanos y animales. La secuencia PEST es el punto de partida para las interacciones proteína-proteína y frecuentemente indican proteínas para degradación (Dramsi 2002; Goldfine 2002).
La Listeriolisina O reconoce el colesterol de membrana, forma poros y favorece la lisis de la membrana del fagolisosoma, emigrando al citosol de la célula hospedero, donde encuentra el medio favorable para la multiplicación (Goebel 2000b; Goldfine 2002).
Una vez la LLO perfora la membrana vacuolar, es reconocida por enzimas en el citoplasma de las células y destruída antes de que pueda dañar la membrana celular (Moors et al., 1999). La LLO actúa como un estímulo inflamatorio induciendo activación celular endotelial, activación neutrofílica y apoptosis, y es responsable de la b-hemólisis en agar sangre (Decatur 2000).
Cepas de L. monocytogenes no productoras de Listeriolisina O, pueden sobrevivir dentro de las vacuolas de los fagocitos, pero no se pueden multiplicar e infectar otras células debido a la incapacidad de escapar de las vacuolas (mutantes avirulentas en ratones) (Portnoy 1994; Sheehan et al., 1994).
Dos tipos de fosfolipasas C sintetizadas por L. monocytogenes, las fosfolipasas C fosfatidilinositol específicas (PI-PLC) de 33KDa, codificadas por el gen plcA y las fosfolipasasas C fosfatidilcolina específicas (PC-PLC) de 29KDa, codificadas por el gen plcB, juegan un papel importante en el poder invasivo; se ha demostrado que las bacterias con mutaciones en los genes que codifican para estas enzimas son menos virulentas en ratones que las cepas silvestres (Bubert et al., 1999; Freitag 1999).
Experimentos con cepas mutantes para el gen plcA demuestran que la PI-PLC ayuda en el escape de la primera vacuola fagocítica de macrófagos derivados de médula ósea en murinos (Camilli et al., 1993; Bannam 1999), y de la línea celular de macrófagos murinos J774 (Wadsworth 1999). La infección de células J774 con cepas silvestres de L. monocytogenes desencadena una vía de señalización en la cual la LLO y la PI-PLC cooperan en el desdoblamiento del fosfatidilinositol (PI), componente estructural de la bicapa lipídica de la célula hospedero, para producir diacilglicerol (DAG). El DAG activa la traslocación a la membrana celular de la proteina kinasa C d (PKC d), implicada en la fosforilación y apertura de los canales de calcio en el primer minuto después de la adición de L. monocytogenes sobre las células J774 (Vázquez andDe Boland 1996; Levin et al., 1997). La movilización de PKC d permite la entrada de calcio en el citoplasma, que en combinación con el DAG produce la traslocación de PKC bII a endosomas tempranos; lo que ocurre según estudios de inmunofluorescencia entre 30 segundos y 3 minutos después de la infección de células J774 con cepas silvestres de L. monocytogenes (Figura 5) (Goldfine 2002; Wadsworth 2002).
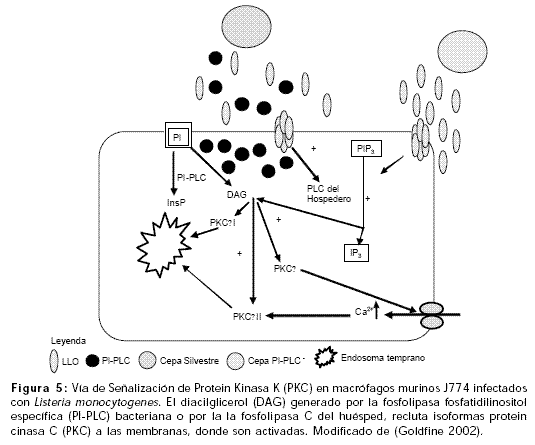
La internalización de cepas silvestres de L. monocytogenes en células J774 genera tres aumentos sucesivos de calcio intracelular que favorecen el escape del microorganismo de la vacuola primaria fagocítica (Wadsworth 1999).
Cepas mutantes de L. monocytogenes que secretan LLO pero no fosfolipasas pueden escindir el PI en DAG y 1, 4, 5-trifosfato de inositol (IP3) a través de las fosfolipasas del hospedero. El DAG induce la traslocación solamente de la PKC bI ya que no hay elevación del calcio intracelular, resultando en un decrecimiento de la eficiencia de escape de las cepas mutantes de la vacuola primaria fagocítica. Estudios de inmunofluorescencia revelan que la traslocación de PKC bI ocurre entre uno a cuatro minutos después de la infección de células J774 con cepas mutantes de L. monocytogenes (Figura 5) (Goldfine 2002; Wadsworth 2002).
El DAG y el calcio son conocidos como activadores de isoformas de PKC en células hospederas infectadas. Las cuatro isoformas de PKC encontradas en macrófagos murinos J774, son a, bI, bII y d. Las isoformas a, bI y bII son activadas por calcio intracelular y/o DAG mientras que la isoforma d es activada por DAG pero es calcio dependiente (Ron 1999; Wadsworth 1999).
La activación de una fosfolipasa eucariótica (PLD) en células J774 coincide con la entrada de la cepa silvestre de L. monocytogenes a la célula hospedero y el comienzo de la maduración vacuolar (Wadsworth 1999).
Una vez que la bacteria ha escapado de la vacuola dentro del citoplasma, se replica gracias a una proteína de superficie con cola hidrofóbica de 60 KDa, enzima hidrolasa p60, también conocida como Iap que cataliza la reacción final de la división celular de L. monocytogenes. La p60 es una proteína modular que contiene dos dominios LysM formados por enzimas involucradas en la degradación de la pared celular, un dominio SH3b homólogo al dominio eucariótico SH3 que media la interacción con las moléculas de señalización del hospedero y un dominio NLPC/P60 (Whisstock 1999; Park 2000).
Análisis de mutaciones en el gen que codifica para p60 indican que es importante para la fagocitosis de L. monocytogenes por algunas lineas celulares como las células intestinales Caco-2 (Kuhn 1999; Park 2000).
C. Nucleación de filamentos de Actina y Expansión célula a célula: Las ATPasa ClpC y ClpE son proteínas de “stress” que ayudan en la disrupción de la membrana vacuolar y la supervivencia intracelular de Listeria. La ClpC modula la expresión de la proteína ActA y de las internalinas a nivel transcripcional (Gaillot et al., 2000).
Para que Listeria monocytogenes pueda moverse directamente a otras células, una proteína de superficie es activada ActA, que está polarmente distribuída sobre la superficie bacteriana e imita la proteína eucariótica WASP (proteína del síndrome de Wiscott-Aldrich) al inducir su propia motilidad dentro del citosol celular (Dramsi 1998; Cossart 2000a).
La ActA puede ser artificialmente dividida en tres regiones: Una región amino terminal cargada positivamente (ActA-N) formada por un dominio ácido (A), un sitio de unión a la actina (AB), una región homóloga al cofilin (C) y un sitio de unión al PI (P); un dominio central hecho de repeticiones ricas en prolina (PRRs); una región carboxi terminal que puede ser suprimida sin afectar el movimiento excepto para la última región de 30 aminoácidos que permite la inserción de la proteína Acta A en la membrana bacteriana (Pistor 1994; Cossart 2001b).
La ActA recluta la proteína VASP (fosfoproteína vasodilatadora) a través del dominio central. La VASP es una proteína encontrada en los sitios activos de polimerización de actina y es un sustrato para las quinasas dependientes de cGMP o cAMP (Niebuhr et al., 1997). La VASP recluta profilin y proporciona monómeros de actina competentes para polimerización a la región ActA-N (Geese et al., 2000). La VASP también interactúa con la actina F a través del dominio carboxi terminal EVH2 formando un puente de unión de la cola a la bacteria (Laurent et al., 1999).
La región ActA-N es esencial para el movimiento de la bacteria (Lasa et al., 1997). La región KKRRK de la ActA-N se une directamente a la actina G y junto con el complejo que contiene siete polipéptidos (complejo Arp2/3) induce la polimerización de moléculas globulares de actina para formar filamentos polarizados de actina (Skoble et al., 2000). Las células bacterianas se mueven a lo largo de estos filamentos hacia la membrana celular y forman estructuras llamadas listeriópodos. Estas protuberancias son fagocitadas por células adyacentes, conduciendo a la diseminación de esta bacteria sin exposición a los anticuerpos (Lasa et al., 1998).
La deleción o mutación de la región KKRRK de la ActA-N impide el reclutamiento del complejo Arp2/3 por la ActA en células infectadas y la polimerización de la actina y el movimiento célula a célula bacteriano (Pistor et al., 2000; Skoble et al., 2000). El complejo Arp2/3 es inactivo “in vitro”, pero cuando es incubado con ActA o con proteínas de la familia WASP/N-WASP/Scar, puede inducir polimerización de la actina (Welch et al., 1998).
La deleción de la región KKRRK en el dominio carboxi-terminal de las proteínas WASP, N-WASP o Scar conducen a la inhibición de la polimerización de la actina e impiden la formación de ondulaciones en la membrana cuando las células son estimuladas por un factor de crecimiento (Rohatgi et al., 1999).
Un tipo novedoso de polimerización de actina inducido por zyxina y el dominio central y carboxi-terminal de la ActA, independiente del complejo Arp2/3 y dependiente de la proteína VASP, ha sido descrito recientemente (Fradelizi et al., 2001). Este tipo de polimerización de actina tiene lugar por contacto focal, y genera más paquetes de filamentos de actina que los filamentos ramificados generados por el complejo Arp2/3, lo que hace pensar que si este fenómeno ocurre in vivo, la ActA podría ser capaz de estimular dos tipos de nucleación de actina: una dependiente del complejo Arp2/3 y otra débilmente dependiente de la proteína VASP (Gouin et al., 1999).
Experimentos con cepas mutantes de L. monocytogenes para el gen plcB demuestran que la PC-PLC es activa durante la expansión célula a célula en murinos (Smith et al., 1995). La PC-PLC causa daño en las membranas vacuolares en algunos tipos de células como las células epiteliales, y pueden ser sustituídas por la LLO (Sibelius et al., 1999).
L. monocytogenes sobrevive dentro de fagosomas secundarios de doble membrana en la nueva célula infectada hasta la disrupción de la membrana vacuolar y la reiniciación de un nuevo ciclo de infección.
Experimentos con cepas de L. monocytogenes no productoras de Listeriolisina O, PC-PLC y SvpA (Lmo2185) (proteína de superficie con cola hidrofóbica similar a la ActA), han demostrado que son requeridas para la lisis del fagosoma secundario formado por la diseminación célula a célula (Gedde et al., 2000; Borezee 2001; Vázquez-Boland et al., 2001a).
En modelos murinos de infección por L. monocytogenes se ha observado un sobrelapamiento funcional de la PI-PLC y PC-PLC, ya que en la ausencia de LLO, la PC-PLC es capaz de mediar el escape de la vacuola primaria en células epiteliales Henle 407 (Marquis et al., 1997).
Otros factores de virulencia que participan en la patogénesis de manera más indirecta, están involucrados en las funciones de mantenimiento y supervivencia del microorganismo al interior del hospedero; funciones necesarias para la vida saprofítica. La p60 (iap) es una proteína extracelular de 60kDa, codificada por el gen iap (gen para la proteína asociada a la invasión). La expresión del gen iap es independiente de PrfA (Bubert et al., 1999), y es controlado a nivel posttranscripcional (Kohler et al., 1991). La proteína p60 que está asociada con la pared celular bacteriana es la responsable de la invasión intestinal y la supervivencia “in vivo” de L. monocytogenes (Hess et al., 1996). Esta proteína tiene una actividad hidrolasa mureína requerida para la formación del septo y esencial para la viabilidad celular (Wuenscher et al., 1993), lo cual hace difícil determinar la función precisa de la p60 en la virulencia debido a que las mutaciones iap son letales; adicionalmente p60, es la proteína de mayor antigenicidad en la respuesta inmune contra L. monocytogenes (Geginat et al., 1999).
Factores antioxidantes: Aunque no es claro como los macrófagos eliminan parásitos intracelulares como L. monocytogenes, se cree que la generación durante la ruptura oxidativa de intermediarios reactivos de oxígeno (ROI), la interacción del anión superóxido y el óxido nítrico (NO) y los intermediarios reactivos de nitrógeno (RNI) realizan un papel importante. Datos experimentales “in vitro” sugieren que ROI y RNI contribuyen significativamente a la muerte de L. monocytogenes mediada por macrófagos (Ohya et al., 1998a; Ohya et al., 1998b). El hierro es requerido para las reacciones catalíticas que conducen a la generación de ROI y RNI, y la correcta homeostasis intracelular de este elemento ha sido reportada como esencial para tener una adecuada actividad listericida de los macrófagos (Fleming 1997).
La catalasa y la superóxido dismutasa (SOD) son enzimas producidas por L. monocytogenes para detoxificar los radicales endógenos de oxígeno libres, generados durante el metabolismo oxidativo, lo cual es importante para contrarestar el mecanismo microbicida dependiente de oxígeno del hospedero, durante la infección (Haas 1993; Miller 1997). Otros mecanismos microbicidas como enzimas lisosomales, defensinas y péptidos antimicrobianos citosólicos son fundamentales para la defensa del hospedero con la infección Listerial (Hiemstra et al., 1999; Shiloh et al., 1999).
Salida de iones metálicos: El hierro es un elemento fundamental para todas las células vivientes, sirve como cofactor para muchas enzimas y está involucrado en el proceso de transporte de electrones. En tejidos animales, el hierro no está disponible libremente porque es secuestrado por la transferrina férrica en el suero y por la ferritina en el componente “hemo” dentro de las células. Así, los patógenos bacterianos tienen que desarrollar mecanismos especializados para capturar el hierro para su crecimiento en tejidos del hospedero (Payne 1993).
El hierro además de estimular el crecimiento de L. monocytogenes en medios sintéticos, incrementa la proliferación bacteriana en hígado y bazo, disminuyendo la dosis letal cincuenta (DL50) (Vázquez-Boland et al., 2001b), siempre y cuando sea administrado en forma salina a ratones infectados. En humanos, la hipersideremia después de transfusiones de sangre masivas ha sido identificada como factor de riesgo importante para la listeriosis (Schuchat et al., 1991).
Tres sistemas de salida de hierro han sido descritos en L. monocytogenes. Uno involucra el transporte directo de citrato férrico a la célula bacteriana por un sistema inducible por citrato (Adams et al., 1990). Otro sistema involucra una reductasa férrica extracelular, la cual usa como sustrato catecolaminas cargadas con hierro y sideróforos (Coulanges et al., 1997; Coulanges et al., 1998). El tercer sistema involucra una proteína de unión a la transferrina localizada en la superficie celular bacteriana (Hartford et al., 1993), aunque la existencia de tal mecanismo ha sido cuestionado (Bhatt et al., 1994). Adicionalmente al papel como nutriente esencial, el hierro es también utilizado por L. monocytogenes como una molécula señal para la regulación de la expresión de los genes de virulencia. Este sistema sensor está basado en la diferencia de concentraciones de hierro libre entre el ambiente y el lumen intestinal (alto) y los tejidos del hospedero (bajo) (Litwin 1993).
Mediadores de respuesta a estrés: Una pequeña proporción de células de L. monocytogenes sobreviven al estrés originado por las condiciones hostiles de la vacuola fagocítica, y alcanzan el citoplasma, donde proliferan y conducen a la diseminación de la infección (De Chastellier 1994). La supervivencia bajo estrés involucra una respuesta adaptativa mediada por un grupo de proteínas conservadas que son reguladas “in vitro” al exponerse a choque térmico, pH bajo, agentes oxidativos, componentes químicos tóxicos y en general cualquier situación en la que el crecimiento bacteriano es afectado. Estas proteínas son chaperonas que contribuyen al plegamiento de proteínas o al ensamblaje de subunidades de proteasas que no pueden ser alteradas conformacionalmente, garantizando la función fisiológica esencial en células bajo condiciones de estrés (Doyle 2001).
Algunas proteínas Clp caseinolíticas, actúan como chaperonas y como enzimas proteolíticas involucradas en la patogénesis de L. monocytogenes (Doyle 2001). Las proteínas Clp son una familia de proteínas de choque térmico altamente conservadas, que están involucradas en la regulación de la proteólisis dependiente de ATP para eliminar proteínas tóxicas desnaturalizadas (Rouquette et al., 1996). La proteína ClpC es requerida para la adhesión y la invasión de L. monocytogenes, modulando la expresión de los genes InlA, InlB y ActA (Nair et al., 2000).
Las ATPasas ClpC y ClpE son requeridas para la supervivencia in vivo y el crecimiento intracelular en macrófagos. Estudios con mutantes dobles clpCclpE en L. monocytogenes han demostrado que estas proteínas contribuyen a la supervivencia intracelular del patógeno (Rouquette et al., 1998; Shen et al., 1998).
En conclusion, en los últimos 20 años, L. monocytogenes ha pasado de ser el agente causal de una enfermedad infecciosa de importancia limitada a uno de los microorganismos zoonóticos típicos transmitidos por alimentos y de mayor interés para las autoridades de salud pública y la industria de alimentos. Es importante el concocimiento sobre parasitismo intracelular practicado por esta bacteria, los determinantes moleculares responsables de las principales etapas de su ciclo de vida dentro de la célula hospedero, la fisiopatología de la listeriosis, manifestaciones clínicas y el tratamiento en las poblaciones de riesgo. Hasta hace poco, los estudios de los determinantes moleculares de L. monocytogenes estaban basados en el análisis de mutantes fenotípicas de este microorganismo, expuestas bajo condiciones in vitro. La tendencia actual es la aplicación de la tecnología genómica en el análisis de la secuencia genómica completa.
El conocimiento acumulado de la inmunobiología de la listeriosis experimental en el ratón, ha hecho de L. monocytogenes un candidato como sistema de producción antigénica para el desarrollo de nuevas vacunas vivas recombinantes. Experimentos recientes han mostrado que este microorganismo puede servir como vector para transferir ADN dentro de células eucarióticas para terapia génica. El progreso en la genómica de L. monocytogenes y el conocimiento de su fisiología puede servir para identificar moléculas blanco para el desarrollo de nuevos agentes antimicrobianos en el tratamiento de listeriosis y otras infecciones bacterianas o sustancias inhibidoras para el control selectivo de la supervivencia y crecimiento de L. monocytogenes en los alimentos.
BIBLIOGRAFÍA
1. Abram M, Doric M. Primary Listeria monocytogenes Infection in Gestating Mice. Folia Microbiol 1997; 42: 65-71. [ Links ]
2. Adams T J, Vartivarian S, Cowart R E. Iron Acquisition System of Listeria monocytogenes. Infect Immun 1990; 58: 2715-2718. [ Links ]
3. Aliprantis A O. Cell Activation and Apoptosis by Bacterial Lipoproteins Through Toll-like Receptor-2. Science 1999; 285: 736-739. [ Links ]
4. Armstrong R W, Fung, P.C. Brainstem Encephalitis Due to Listeria monocytogenes: Case Report and Review. Clin Infect Dis 1993; 16: 689-702. [ Links ]
5. Bannam T, Goldfine, H. Mutagenesis of Active-site Histidines of Listeria monocytogenes Phosphatidylinositol-specific Phospholipase C: Effects on Enzyme Activity and Biological Function. Infect Inmun 1999; 67: 182-186. [ Links ]
6. Bartfield A. Bacterial Meningitis. Obstetric and Gynecology 2000; 7(2): 49-54. [ Links ]
7. Becroft K F. Epidemic Listeriosis in the Newborn. British Med J 1971; 3: 747-751. [ Links ]
8. Bhatt R, AlaAldeen D A, Baldwin T, Boriello S P. The 126 KDa Iron Regulated Protein of Listeria monocytogenes is Not a Transferrin Binding Protein. FEMS Microbiol Let 1994; 123: 119-123. [ Links ]
9. Bierne H. A Role for Cofilin and LIM Kinase in Listeria-induced Phagocytosis. J Cell Biol 2001; 155: 101-112. [ Links ]
10. Bierne H, Cossart P. InlB, a Surface Protein of Listeria monocytogenes that Behaves as an Invasin and a Growth Factor. J Cell Sci 2002; 115: 3357-3367. [ Links ]
11. Bille J. Epidemiology of Human Listeriosis in Europe, with Special Reference to the Swiss Outbreak. Miller A J, Smith, J.L., Somkuti, G.A. Foodborne Listeriosis. New York, 1990. Elsevier: 71-74. [ Links ]
12. Blanot S, Joly M M, Vilde F. A Gerbil Model for Rhomboencephalitis Due to Listeria monocytogenes. Microb Path 1997; 23: 39-48. [ Links ]
13. Borezee E. SvpA, a Novel surface Virulence-associated Protein Required for Intracellular Survival of Listeria monocytogenes. Microbiol 2001; 147: 2913-2923. [ Links ]
14. Brosch R, Catimel B, Milon G, Buchrieser C, Vindel ERocourt J. Virulence Heterogencity of Listeria monocytogenes Strains from Various Sources (Food, Human, Animal) in Inmunocompetent Mice and Its Association With Typing Characteristics. J Food Prot 1993; 56: 296-301. [ Links ]
15. Bubert A, Sokolovic Z, Chun S K, Papatheodorou L, Simm AGoebel W. Differential Expression of Listeria monocytogenes Virulence Genes in Mammalian Host Cells. Mol Gen Genet 1999; 261: 323-336. [ Links ]
16. Cabanes D, Dehoux P, Dussurget O, Frangeul LCossart P. Surface Proteins and the Pathogenic Potential of Listeria monocytogenes. TRENDS Microbiol 2002; 10(5): 238-245. [ Links ]
17. Camilli A, Tilney L G, Portnoy D A. Dual Roles of plcA in Listeria monocytogenes Pathogenesis. Mol Microbiol 1993; 8: 143-157. [ Links ]
18. Carvajal A, Samuelsson S, Rothgardt N PFredeiksen W. The Treatment of Listeria monocytogenes Infection in the Central Nervous System. Acta Microbiol Hung1989; 36: 159-163. [ Links ]
19. Chakraborty T, Hain T, Domann E. Genome Organization and the Evolution of the Virulence Gene Locus in Listeria Species. International J Med Microbiol 2000; 290: 167-174. [ Links ]
20. Charpentier E, Courvalin P. Emergence of the Trimethoprim Resistance Gene dfrD in Listeria monocytogenes BM 4293. Antimicrob Agents Chemother 1997; 41: 1134-1136. [ Links ]
21. Cherubin C E, Appleman M D, Heseltime P N R, Khayr WStratton C W. Epidemiological Spectrum and Current Treatment of Listeriosis. Rev Infect Dis 1991; 13: 1108-1114. [ Links ]
22. Conlan J C, North R J. Neutrophil-mediated Dissolution of Infected Host Cells as a Defense Strategy Against a Facultative Intracellular. J Exp Med 1991; 174: 741-744. [ Links ]
23. Conlan J W, North R J. Early Pathogenesis of Infection in the Liver with the Facultative Intracellular Bacteria Listeria monocytogenes, Francisella tularensis and Salmonella Typhimurium Involves Lysis of Infected Hepatocytes by Leukocytes. Infec Immun 1992; 60: 5164-5171. [ Links ]
24. Cossart P. Actin-based Motility of Pathogens: the Arp2/3 Complex is a Central Player. Cell Microbiol 2000a; 2: 195-205. [ Links ]
25. Cossart P. Met, The HGF-SF Receptor: Another Receptor for Listeria monocytogenes. Trends Microbiol 2001a; 9(3): 105-107. [ Links ]
26. Cossart P, Bierne, H. The Use of Host Cell Machinery in the Pathogenesis of Listeria monocytogenes. Curr Opin Inmunol 2001b; 13: 96-103. [ Links ]
27. Cossart P, Jonquieres, R. Sortase, a Universal Target for Therapeutic Agents Against Gram Positive Bacteria? 2000b. Proc Nat Acad Sci, USA. 5013-5015. [ Links ]
28. Cossart P, Pizarro-Cerdá JLecuit M. Invasion of Mammalian Cells by Listeria monocytogenes: Functional Mimicry to Subvert Cellular Functions. TRENDS Cell Biolo 2003; 13(1): 23-31. [ Links ]
29. Cossart P, Portnoy, D. Gram-Positive Pathogens. 2001c. Washington, American Society for Microbiology. 507-515. [ Links ]
30. Coulanges V, Andre PVidon D J. Effects of Siderophores, Catecholamines, and Catechol Compounds on Listeria spp. Growth in Iron-complexed Medium. Biochem Biophys Res Commun 1998; 249: 526-530. [ Links ]
31. Coulanges V, Andre P, Ziegler O, Buchheit LVidon D J. Utilization of Iron Catecholamine Complexes Involving Ferric Reductase Activity in Listeria monocytogenes. Infect Immun 1997; 65: 2778-2785. [ Links ]
32. Cousens L P, Wing E J. Innate Defenses in the Liver During Listeria Infection. Inmunol Rev 2000; 174: 150-159. [ Links ]
33. Dalton C B, Austin C, Sobel J, Hayes P, Bibb W, Graves L, Swaminathan B, Proctor MGriffin P M. An Outbreak of Gastroenteritis and Fever Due to Listeria monocytogenes in Milk. New England J Med 1997; 336(2): 100-105. [ Links ]
34. De Chastellier C, Berche, P. Fate of Listeria monocytogenes in Murine Macrophages: Evidence for Simultaneous Killing and Survival of Intracellular Bacteria. Infect Immun 1994; 62: 543-553. [ Links ]
35. Decatur A L, Portnoy, D.A. A PEST-like Sequence in Listeriolysin O Essential for Listeria monocytogenes Pathogenicity. Science 2000; 290: 992-995. [ Links ]
36. Dhar G. Anchor Structure of Cell Wall Surface Proteins in Listeria monocytogenes. Biochem 2000; 39: 3725-3733. [ Links ]
37. DiMaio H. Listeria Infection in Women. Obst Ginecol 2000; 7: 40-45. [ Links ]
38. Doyle M E. Virulence Characteristics of Listeria monocytogenes. FRI Brief 2001: 1-13. [ Links ]
39. Dramsi S, Biswas I, Maguin E, Braun L, Mastroeni PCossart P. Entry of Listeria monocytogenes Into Hepatocytes Requires the Expression of InlB, a Surface Protein of the Internalin Multigene Family. Mol Microbiol 1995; 16: 251-261. [ Links ]
40. Dramsi S, Cossart P. Intracelullar Pathogens and the Actin Cytoskeleton. Ann Rev Cell and Develop Biol 1998; 14: 137-166. [ Links ]
41. Dramsi S, Cossart, P. Listeriolysin O: a Genuine Cytolysin Optimized for an Intracellular Parasite. J Cell Biol 2002; 156: 943-946. [ Links ]
42. Drevets D A. Dissemination of Listeria monocytogenes by Infected Phagocytes. Infect Inmun 1999; 67(7): 3512-3517. [ Links ]
43. Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, Mukaida NNaito M. The Role of Kupffer Cells and Regulation of Neutrophil Migration Into the Liver by Macrophage Inflammatory Protein-2 in Primary Listeriosis in Mice. Pathol Int 1999; 49: 519-532. [ Links ]
44. Edelson B, Cossart P, Unanue R. Paradigm Revisited: Antibody Provides Resistance to Listeria Infection. J Inmunol 1999; 163: 4087-4090. [ Links ]
45. Eden S. Mechanism of Regulation of WAVE1-induced Actin Nucleation by Rac1 y Nck. Nature 2002; 418: 790-793. [ Links ]
46. Emoto Y, Emoto M, Kaufmann S H E. Transient Control of Interleukin-4 Producing Natural Killer T Cells in the Livers of Listeria monocytogenes Infected Mice by Interleukin-12. Infect Immun 1997; 65: 5003-5009. [ Links ]
47. Espaze E P, Rocourt J, Courtieu A L. La Listerióse en France en 1989. Etude à Partir des Souches Adressées au Centre National de Référence. Bulletin Epidémiologie Hebdomaire 1991; 3: 9-10. [ Links ]
48. Evans J R, Allen A C, Bortolussi R, Issekutz T B, Stinson D A. Follow-up Study of Survivors of Fetal and Early-onset Neonatal Listeriosis. Clin Invest Med 1984; 7: 329-334. [ Links ]
49. Finlay B B, Cossart P. Exploitation of Mammalian Host Cell Functions by Bacterial Pathogens. Science 1997; 276: 718-725. [ Links ]
50. Fleming S D, Campbell, P.A. Some Macrophages Kill Listeria monocytogenes While Others Do Not. Inmunol Rev 1997; 158: 69-77. [ Links ]
51. Fradelizi J, Noireaux V, Plastino J, Menichi B, Louvard D, Sykes C, Golsteyn RFriederich E. ActA and Human Zyxin Harbour Arp2/3-independent Actin-polimerization Activity. Nat Cell Biol 2001; 3(8): 699-707. [ Links ]
52. Freitag N E, Jacobs K E. Examination of Listeria monocytogenes Intracellular Gene Expression by Using the Green Fluorescent Protein of Aequorea victoria. Infect Immun 1999; 67: 1844-1852. [ Links ]
53. Gaillard J L. Entry of L. monocytogenes into Cells is Mediated by Internalin, a Repeat Protein Reminiscent of Surface Antigens from Gram-positive Cocci. The Cell 1991; 65: 1127-1141. [ Links ]
54. Gaillard J L, Jaubert F, Berche P. The inlAB Locus Mediates The Entry of Listeria monocytogenes Into Hepatocytes In Vivo. J Exp Med 1996; 183: 359-369. [ Links ]
55. Gaillot O, Pellegrini E, Bregenholt S, Nair SBerche P. The ClpP Serine Protease in Essential for the Intracellular Parasitism and Virulence of Listeria monocytogenes. Mol Microbiol 2000; 35: 1286-1294. [ Links ]
56. Gedde M M, Higgins D E, Tilney L GPortnoy D A. Role of Listeriolysin O in Cell-to-Cell Spread of Listeria monocytogenes. Infect Inmun 2000; 68(2): 999-1003. [ Links ]
57. Geese M, Schluter K, Rothkegel M, Jockusch B M, Wehland JSechi A S. Accumulation of Profilin II at the Surface of Listeria is Concomitant with the Onset of Motility and Correlates with Bacterial Speed. J Cell Sci 2000; 113: 1415-1426. [ Links ]
58. Geginat G, Lalic M, Kretschmar M, Goebel W, Hof H, Palm DBubert A. Th1 Cells Specific for a Secreted Protein of Listeria monocytogenes Are Protective in Vivo. J Inmunol1998; 160: 6046-6055. [ Links ]
59. Geginat G, Nichterlein T, Kretschmar M, Schenk S, Hof H, Lalic M, Goebel WBubert A. Enhancement of the Listeria monocytogenes p60 Specific CD4 and CD8 T Cell Memory by Nonpathogenic Listeria innocua. J Inmunol 1999; 162: 4781-4789. [ Links ]
60. Gilot P, André P, Content J. Listeria monocytogenes Possesses Adhesins for Fibronectin. Infect Inmun 1999; 67(12): 6698-6701. [ Links ]
61. Goebel W, Kreft J, Bockmann R. Gram-Positive Pathogens. 2000a. Washington, American Society for Microbiology. 499-506. [ Links ]
62. Goebel W, Kuhn M. Bacterial Replication in the Host Cell Cytosol. Curr Opin Microbiol 2000b; 3: 49-53. [ Links ]
63. Goldfine H, Wadsworth S. Macrophage Intracellular Signaling Induced by Listeria monocytogenes. Microb Infect 2002; 4: 1335-1343. [ Links ]
64. Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti P J, Cossart P. A Comparative Study of the Actin-based Motilities of the Pathogenic Bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J Cell Science 1999; 112: 1697-1708. [ Links ]
65. Goulet V, Lepoutre A, Rocourt J, Courtieu A L, Dehaumont P, Veit P. L’épidémie de Listériose en France. Bilan Final et Résultats de L’enquète Épidémiologique. Bull Epidém Hebdo 1993; 4: 13-14. [ Links ]
66. Graham J, Lanser S, Bignardi G, Pedler SHollyoak V. Hospital Acquired Listeriosis. J Hosp Infect 2002; 51: 136-139. [ Links ]
67. Gray M L, Killinger A H. Listeria monocytogenes and Listeric Infections. Bacteriol Rev 1966; 30: 309-382. [ Links ]
68. Gregory S H, Liu C C. CD8+ T Cell Mediated Response to Listeria monocytogenes Taken Up in the Liver and Replicating Within Hepatocytes. Inmunol Rev 2000; 174: 112-122. [ Links ]
69. Gregory S H, Wing E J. Accessory Function of Kupffer Cells in the Antigen Especific Blastogenic Response of an L3T4+ T-lymphocyte Clone to Listeria monocytogenes. Infect Immun 1990; 58: 2313-2319. [ Links ]
70. Greiffenberg L, Goebel W, Kim K S, Daniels J, Kuhn M. Interaction of Listeria monocytogenes With Human Brain Microvascular Endothelial Cells: An Electron Microscopic Study. Infect Immun 2000; 68: 3275-3279. [ Links ]
71. Guleria I, Pollard, J.W. The Trophoblast is a Component of the Innate Inmune System During Pregnancy. Nature Medicine 2000; 6: 589-593. [ Links ]
72. Haas A, Brehm K. Superoxide Dismutases and Catalases Biochemistry, Molecular Biology and Some Biomedical Aspects. Genet Eng Biotechnol 1993; 13: 243-269. [ Links ]
73. Hacker J, Kaper J. The Concept of Pathogenicity Islands. 2000. Pathogenicity Islands of Virulent Bacteria: Structure, Function, and Impact on Microbial Evolution. Washington, American Society for Microbiology: 1-12. [ Links ]
74. Hartford T, O’Brien S, Andrew P W, Jones D, Roberts I S. Utilization of Transferrin-bound Iron by Listeria monocytogenes. FEMS Microbiol Lett 1993; 108: 311-318. [ Links ]
75. Hauf N, Goebel W, Fiedler F, Sokolovic Z, Kuhn M. Listeria monocytogenes Infection of P388D1 Macrophages Results in a Biphasic NF-KB (RelA/p50) Activation Induced by Lipoteichoic Acid and Bacterial Phospholipases and Mediated by IkBa and IkBb Degradation. 1997. Proc Nat Acad Sci, USA. 9394-9399. [ Links ]
76. Hauf N, Goebel W, Serfling E, Kuhn M. Listeria monocytogenes Infection Enhances Transcription Factor NF-kappab in P388D1 Macrophage -like Cells. Infect Immun 1994; 62: 2740-2747. [ Links ]
77. Hermiston M L. Forced Expression of E-cadherin in the Mouse Intestinal Epithelium Slows Cell Migration and Provides Evidence for Nonautonomous Regulation of Cell Fate in a Self-renewing System. Gen & Develop 1996; 10: 985-996. [ Links ]
78. Hess J, Dreher A, Gentschev I, Goebel W, Ladel C, Miko DKaufmann S H. Protein p60 Participates in Intestinal Host Invasion by Listeria monocytogenes. Zentralbl Bakteriol 1996; 284: 263-272. [ Links ]
79. Hiemstra P S, Van den Barselaar M T, Roest M, Nibbering P H, Van Furth R. Ubiquicidin, a Novel Murine Microbicidal Protein Present in the Cytosolic Fraction of Macrophages. J Leuk Biol 1999; 66: 423-428. [ Links ]
80. Hof H, Nichterlein T, Kretschmar M. Management of Listeriosis. Clin Microbiol Rev 1997; 10(2): 345-357. [ Links ]
81. Hofer E, Oliveira L M A. Sensibilidade Antimicrobiana em Amostras de Listeria Isoladas de Diferentes Fontes e Regioes do Brasil. Reva Microbiol, Sao Paulo 1988; 19: 109-112. [ Links ]
82. Ireton K, Payrastre B, Chap H, Ogawa W, Sakave H, Kasuga M, Cassart P. A Role for Phosphoinositide 3 - kinase in Bacterial Invasion. Science 1996; 274: 780-781. [ Links ]
83. Jacquet C, Catimel B, Goulet V, Lepoutre A, Veit P, Dehaumont P, Rocourt J. Typing of Listeria monocytogenes During Epidemiological Investigations of the French Listeriosis Outbreaks in 1992, 1993 and 1995. Proceedings of the 12th International Symposium on Problems of Listeriosis. Promaco Conventions. Perth, Western Australia: 161-176. [ Links ]
84. Jensen V B, Harty J T, Jones B D. Interactions of the Invasive Pathogens Salmonella Typhimurium, Listeria monocytogenes, and Shigella flexneri with M Cells and Murine Peyer’s Patches. Infect Inmun 1998; 66: 3758-3766. [ Links ]
85. Jonquieres R. Interaction Between the Protein InlB of Listeria monocytogenes and Lipoteichoic Acid: a Novel Mechanism of Protein Association at the Surface of Gram-positive. Mol Microbiol 1999; 34: 902-914. [ Links ]
86. Jonquieres R. Synergy Between the N-and C-terminal Domains of InlB for Efficient Invasion of Non-phagocytic Cells by Listeria monocytogenes. Mol Microbiol 2001; 42: 955-965. [ Links ]
87. Jungi T W, Pfister H, Sager H, Fatzer R, Vandevelde M, Zurbriggen A. Comparison of Inducible Nitric Oxide Synthase Expression in the Brains of Listeria monocytogenes Infected Cattle, Sheep, and Goats and in Macrophages Stimulated in Vitro. Infect Inmun 1997; 65: 5279-5288. [ Links ]
88. Kajava A V. Structural Diversity of Leucine-rich Repeat Proteins. Journal of Molecular Biology 1998; 277: 519-527. [ Links ]
89. Kalstone C. Successful Antepartum Treatment of Listeriosis. Amer J Obst Gynecol 1991; 164: 57-58. [ Links ]
90. Kayal S, Lilienbaum A, Poyart C, Memet S, Israel A, Berche P. Listeriolysin O-dependent Activation of Endothelial Cells During Infection with Listeria monocytogenes: Activation of NF-kB and Upregulation of Adhesion Molecules and Chemokines. Mol Microbiol 1999; 31: 1709-1722. [ Links ]
91. Kendall M J, Clarke S WSmith W T. Spinal Abscess Due to Listeria monocytogenes in a Patient with Hepatic Cirrhosis. J Pathol 1972; 107: 9-11. [ Links ]
92. Klatt E C, Pavlova Z, Teberg A J, Yonekura M L. Epidemic Neonatal Listeriosis at Autopsy. Hum Pathol 1986; 17: 1278-1281. [ Links ]
93. Kohler S, Bubert A, Vogel M, Goebel W. Expression of the iap Gene Coding for Protein p60 of Listeria monocytogenes is Controlled on the Posttranscriptional Level. J Bacteriol 1991; 173: 4668-4674. [ Links ]
94. Kuhn M, Goebel W. Pathogenesis of Listeria monocytogenes. 1999. Listeria, Listeriosis and Food Safety. Ryser E T, Marth E H, Dekker M. New York: 97-130.
95. Kussel-Andermann P. Vezatin, a Novel Transmembrane Protein, Bridges Myosin VIIA to the Cadherin-catenins Complex. Eur Mol Biol Org J 2000; 19: 6020-6029. [ Links ]
96. Lasa I, Dehoux P, Cossart P. Actin Polymerization and Bacterial Movement. Biochim Biophys Acta 1998; 1402: 217-228. [ Links ]
97. Lasa I, Gouin E, Goethals M, Vancompernollle K, David V, Vandekerckhove J, Cossart P. Identification of Two Regions in the Amino-terminal Domain of ActA Involved in the Actin Comet tail Formation by Listeria monocytogenes. European Mol Biol Org J 1997; 16: 1531-1540. [ Links ]
98. Laurent V, Loisel T, Harbeck B, Wehman A, Grobe L, Jockusch B M, Wehland J, Gertler F B, Carlier M F. Role of Proteins of the Ena/VASP Family in Actin-based Motility of Listeria monocytogenes. J Cell Biol 1999; 144: 1245-1258. [ Links ]
99. Lavon I, Goldberg I, Amit S, Landssman I, Jung S, Tsuberi B Z, Barshack I, Kopolovic J, Galun E, Bujard H, Ben-Neriah Y. High Susceptibility to Bacterial Infection, But No Liver Disfunction, in Mice Compromised for Hepatocyte NF-kappaB Activation. Nat Med 2000; 6: 573-577. [ Links ]
100. Lebrun M, Audurier A, Cossart P. Plasmid-borne Cadmiun Resistance Genes in Listeria monocytogenes Are Similar to cadA and cadC of Staphylococcus aureus and Are Induced by Cadmium. J Bacteriol 1994; 176: 3040-3048. [ Links ]
101. Lecuit M. A Single Amino Acid in E-cadherin Responsible for Host Specificity Towards the Human Pathogen Listeria monocytogenes. Eur Mol Biol Org J 1999; 18: 3956-3963. [ Links ]
102. Lecuit M. A Role for a and b Catenins in Bacterial Uptake. 2000. Proc Nat Acad Sci, USA. 10008-10013. [ Links ]
103. Lecuit M. A Transgenic Model for Listeriosis; Role of Internalin in Crossing the Intestinal Barrier. Science 2001; 292: 1722-1725. [ Links ]
104. Lecuit M, Cossart, P. Genetically-modified-animal Models for Human Infections; the Listeria Paradigm. TRENDS Mol Med 2002; 8(11): 537-542. [ Links ]
105. Leimeister-Wachter M, Domann E, Chakraborty T. The Expression of Virulence Genes in Listeria monocytogenes is Thermoregulated. J Bacteriol 1992; 174: 947-952. [ Links ]
106. Levin R, Braiman A, Priel Z. Protein Kinase C Induced Calcium Influx and Sustained Enhancement of Ciliary Beating by Extracellular ATP. Cell Calcium 1997; 21: 103-113. [ Links ]
107. Linnan M J, Mascola L, Lou X D, Goulet V, May S, Salminen R N, Hird D W, Yonekura M L, Hayes P, Weaver R, Audurier A, Plikaytis B D, Fannin S L, Fleks A, Broome C V. Epidemic Listeriosis Associated with Mexican-style Cheese. New Eng J of Med 1988; 319: 823-828. [ Links ]
108. Litwin C M, Calderwood S B. Role of Iron in Regulation of Virulence Genes. Clin Microbiol Rev 1993; 6: 137-149. [ Links ]
109. López S, Marco A J, Prats N, Czuprynski C J. Critical Role of Neutrophils in Eliminating Listeria monocytogenes From the Central Nervous System During Experimental Murine Listeriosis. Infect Immun 2000; 68: 4789-4791. [ Links ]
110. Lorber B. Listeriosis. Clin Infect Dis 1996; 24: 1-11. [ Links ]
111. Low J C, Donachie, W. Clinical and Serum Antibody Responses in Lambs to Infection by Listeria monocytogenes. Res Vet Sci 1991; 51: 185-192. [ Links ]
112. Low J C, Donachie W. A Review of Listeria monocytogenes and Listeriosis. Vet J 1997; 153: 9-29. [ Links ]
113. Low J C, Wright F, McLauchlin J, Donachie W. Serotyping and Distribution of Listeria Isolates From Cases of Ovine Listeriosis. Vet Rec 1993; 133: 165-166. [ Links ]
114. Machesky L, May R C. Phagocytosis and the Actin Cytoskeleton. J Cell Sci 2001; 114: 1061-1077. [ Links ]
115. Mackaness G B, Hill W C. The Effect of Anti-lymphocyte Globulin on Cell-mediated Resistance to Infection. J Exp Med 1969; 129: 993-1012. [ Links ]
116. Mansell A. Internalin B Activates Nuclear Factor-kB Via Ras, Phosphoinositide 3-kinase, and Akt. J Biol Chem 2001; 276: 43597-43603. [ Links ]
117. Marco A J, Altimira J, Prats N, López S, Domínguez L, Domingo M, Briones V. Penetration of Listeria monocytogenes in Mice Infected by the Oral Route. Microb Pathol 1997; 23: 255-263. [ Links ]
118. Marco A J, Prats N, Ramos J A, Briones V, Blanco M, Domínguez LDomingo M. A Microbiological, Histopathological and Inmunohistological Study of the Intragastric Inoculation of Listeria monocytogenes in Mice. J Comp Pathol 1992; 107: 1-9. [ Links ]
119. Marino M. GW Domains of the Listeria monocytogenes Invasion Protein InlB are SH3-like and Mediate Binding to Host Ligands. Eur Mol Biol Org J 2002; 21: 5623-5634. [ Links ]
120. Marquis H, Goldfine H, Portnoy D A. Proteolytic Pathways of Activation and Degradation of a Bacterial Phospholipase C During Intracellular Infection by Listeria monocytogenes. J Cell Biol 1997; 137: 1381-1392. [ Links ]
121. Marth E H. Disease Characteristics of Listeria monocytogenes. Food Technol 1988; 42: 165-168. [ Links ]
122. McLauchin J. The Relationship Between Listeria and Listeriosis. Food Cont 1996; 7(4): 187-193. [ Links ]
123. McLauchlin J. Distribution of Serovars of Listeria monocytogenes Isolated From Different Categories of Patients With Listeriosis. Eur J Clin Microbiol Infect Dis 1990; 9: 210-213. [ Links ]
124. McLauchlin J, Hall S M, Velani S K, Gilbert R J. Human Listeriosis and Paté: a Possible Association. British Med J 1991; 303: 773-775. [ Links ]
125. Menudier A, Basiraud C, Jean - Albert N. Virulence of L. monocytogenes Serovars and Listeria spp. in Experimental Infection of Mice. J Food Prot 1991; 54(12): 917-921. [ Links ]
126. Miller R, Britigan, B.E. Role of Oxidants in Microbial Pathophysiology. Clin Microbiol Rev 1997; 10: 1-18. [ Links ]
127. Moellering R C, Medoff G, Leech I, Wennersten C, Kunz L J. Antibiotic Synergism Againts Listeria monocytogenes. Antimicrob Agents Chemoth 1972; 1: 30-34. [ Links ]
128. Moors M A, Levitt B, Youngman P, Portnoy D A. Expression of Listeriolysin O and ActA by Intracellular and Extracellular Listeria monocytogenes. Infect Inmun 1999; 67: 131-139. [ Links ]
129. Morritt A, Mclean N, Snow M. Oral Cancer, Fever of Unknown Origin, and Listeriosis. British J Oral Maxillo Surg 2002; 40: 442-443. [ Links ]
130. Mrowka M, Graf L P, Odin P. MRI Findings in Mesenrhombencephalitis Due to Listeria monocytogenes. J Neurol Neurosurg & Psych 2002; 73: 775. [ Links ]
131. Nagafuchi A. Molecular Architecture of Adherens Junctions. Curr Opin Cell Biol 2001; 13: 600-603. [ Links ]
132. Nair S, Milohanic E, Berche P. ClpC ATPase is Required for Cell Adhesion and Invasion of Listeria monocytogenes. Infect Inmun 2000; 68: 7061-7068. [ Links ]
133. Nakane A, Minagawa T, Yasuda I. Induction of Alpha/beta Interferon and Gamma Interferon in Mice Infected With Listeria monocytogenes During Pregnancy. Infect Immun 1985; 50: 877-880. [ Links ]
134. Niebuhr K, Ebel F, Frank R, Reinhard M, Domann E, Carl U D, Walter U, Gertler F B, Wehland JChakraborty T. A Novel Proline-rich Motif Present in ActA of Listeria monocytogenes and Cytoskeletal Proteins is the Ligand for the EVH1 Domain, a Protein Module Present in the Ena/VASP Family. European Molecular Biology Organization Journal 1997; 16: 5433-5444. [ Links ]
135. Nivia S, Zipf ABhunia A. Influence of Temperature and Growth Phase on Expression of a 104- Kilodalton Listeria Adhesion Protein in Listeria monocytogenes. App Env Microbiol 1999; 65: 2765-2769. [ Links ]
136. Nolla-Salas J, Almela M, Passer I, Latorre C, Salvadó M, Coll P. Spontaneous Listeria monocytogenes Peritonitis: A Population Based Study of 13 Cases Collected in Spain. American J Gastroenterol 2002; 97(6): 1507-1511. [ Links ]
137. Norrung B, Andersen J K. Variations in Virulence Between Different Electrophoretic Types of Listeria monocytogenes. Lett App Microbiol 2000; 30: 228-232. [ Links ]
138. North R J. The Concept of the Activated Macrophage. J Inmunol 1978; 121: 806-809. [ Links ]
139. Ohya S, Tanabe Y, Makino M, Nomura T, Xiong H, Arakawa M, Mitsuyama M. The Contributions of Reactive Oxygen Intermediates and Reactive Nitrogen Intermediates to Listericidal Mechanisms Differ in Macrophages Activated pre- and Postinfection. Infect Inmun 1998a; 66: 4043-4049. [ Links ]
140. Ohya S, Xiong H, Tanabe Y, Arakawa M, Mitsuyama M. Killing Mechanisms of Listeria monocytogenes in Activated Macrophages as Determinated by an Improved Assay System. J Med Microbiol 1998b; 47: 211-215. [ Links ]
141. Pandiripally V K, Westbrook D G, Sunki G R, Bhunia A K. Surface Protein p104 is Involved in Adhesion of Listeria monocytogenes to Human Intestinal Cell Line, Caco-2. J Med Microbiol 1999; 48: 117-124. [ Links ]
142. Parham P, Unanue E R. Inmunity to L. monocytogenes: A Model Intracelullar Pathogen. Inmunol Rev 1997; 158: 1-169. [ Links ]
143. Park J H. Specific Binding of Recombinant Listeria monocytogenes p60 Protein to Caco-2 Cells. FEMS Microbiol Lett 2000; 186: 35-40. [ Links ]
144. Parkassh V, Morotti R A, Joshi V, Cartun R, Rauch C A, West A B. Inmunohistochemical Detection of Listeria Antigen in the Placenta in Perinatal Listeriosis. Int J Gyn Pathol 1998; 17: 343-350. [ Links ]
145. Patel R, Paya C V. Infections in Solid-organ Transplant Recipients. Clin Microbiol Rev 1997; 10: 86-124. [ Links ]
146. Payne S M. Iron Acquisition in Microbial Pathogenesis. Trends Microbiol 1993; 1: 66-69. [ Links ]
147. Pinner R W, Schuchat A, Swaminathan B, Hayes P, Deaver K, Weaver R, Plikaytis B, Reeves M, Broome C, Wenger J. Role of Foods in Sporadic Listeriosis II. Microbiologic and Epidemiologic Investigation. J Amer Med Assoc. 1992; 267: 2046-2050. [ Links ]
148. Pistor S. The ActA Protein of Listeria monocytogenes Acts as a Nucleator Inducing Reorganization of the Actin Cytoskeleton. Eur Mol Biol Org J 1994; 13: 758-763. [ Links ]
149. Pistor S, Grobe L, Sechi A S, Dommannn E, Gerstel B, Machesky L, Chakraborty T, Wehland J. Mutations of Arginine Residues Within the 146-KKRRK-150 Motif of the ActA Protein of Listeria monocytogenes Abolish Intracellular Motility by Interfering with the Recruitment of the Arp2/3 Complex. J Cell Sci 2000; 113: 3277-3287. [ Links ]
150. Portnoy D A. Innate Inmunity to a Facultative Intracelullar Bacterial Pathogen. Curr Opi Inmunol 1992; 4: 20-24. [ Links ]
151. Portnoy D A. Molecular Genetics of Bacterial Pathogenesis. 1994. Washington, American Society for Microbiology. 279-293. [ Links ]
152. Portnoy D A, Chakraborty T, Goebel W, Cossart P. Molecular Determinants of Listeria monocytogenes Pathogenesis. Infect Immun 1992; 60: 1263-1267. [ Links ]
153. Poyart-Salmeron C, Carlier C, Trieu-Cuot P, Courtieu A L, Courvalin P. Transferable Plasmid-mediated Antibiotic Resistance in Listeria monocytogenes. The Lancet 1990; 335: 1422-1426. [ Links ]
154. Pron B, Boumaila C, Jaubert F, Sarnacki S, Monnet J P, Berche P, Gaillard J L. Comprehensive Study of the Intestinal Stage of Listeriosis in a Rat Ligated Ileal Loop System. Infect Inmun 1998; 66: 747-755. [ Links ]
155. Rácz P, Tenner K, Méro E. Experimental Listeria Enteritis. I. An Electron Microscopic Study of the Epithelial Phase in Experimental Listeria Infection. Lab Invest 1972; 26: 694-700. [ Links ]
156. Raffelsbauer D. The Gene Cluster inlC2DE of Listeria monocytogenes Contains Additional New Internalin Genes and is Important for Virulence in Mice. Mol Gen Genetics 1998; 260: 144-158. [ Links ]
157. Rasmussen O F, Skouboe P, Dons I, Rossen L, Olsen J E. Listeria monocytogenes Exists in at Least Three Evolutionary Lines: Evidence From Flagellin, Invasive Associated Protein and Listeriolysin O Genes. Microbiol 1995; 141: 2053-2061. [ Links ]
158. Renzoni A, Cossart P, Dramsi S. PrfA, the Transcripcional Activator of Virulence Genes, is Upregulated During Interaction of Listeria monocytogenes with Mammalian Cells and in Eukaryotic Cell Extracts. Mol Microbiol 1999; 34: 552-561. [ Links ]
159. Riedo F X, Pinner R W, Tosca M D, Cartter M L, Graves L M, Broome C V. A Point-source Food-borne Listeriosis Outbreak: Documented Incubation Period and Possible Mild Illness. J Infect Dis 1994; 170: 693-696. [ Links ]
160. Ripio M T, Domínguez-Bernal G, Suárez M, Brehm K, Berche P, Vázquez-Boland J A. Transcriptional Activation of Virulence Genes in Wild-type Strains of Listeria monocytogenes in Response to a Change in the Extracelullar Medium Composition. Res Microbiol 1996; 147: 311-384. [ Links ]
161. Rocourt J. Listeria monocytogenes: The State of the Art. Dairy Food and Env Sanit 1994a; 14: 70-82. [ Links ]
162. Rocourt J. Risk Factors for Listeriosis. Food Control 1996; 7(4): 195-202. [ Links ]
163. Rocourt J, Jacquet C. Epidémiologie des Infections Humanies à Listeria monocytogenes en 1994: Certitudes et Interrogations. Annual Institute Pasteur/Actualités 1994b; 5: 168-174. [ Links ]
164. Rogers H W, Callery M P, Deck B, Unanue E R. Listeria monocytogenes Induces Apoptosis of Infected Hepatocytes. J Inmunol 1996; 156: 679-684. [ Links ]
165. Rogers H W, Unanue E R. Neutrophils Are Involved in Acute, Non-specific Resistance to Listeria monocytogenes in Mice. Infect Immun 1993; 61: 5090-5096. [ Links ]
166. Rohatgi R, Ma L, Miki H, López M, Kirchhausen T, Takenawa T, Kirschner M W. The Interaction Between N-WASP and the Arp2/3 Complex Links Cdc42-dependent Signals to Actin Assembly. The Cell 1999; 97: 221-231. [ Links ]
167. Ron D, Kazanietz M G. New Insights Into the Regulation of Protein Kinase C and Novel Phorbol Ester Receptors. Fed Amer Soc Exp Biol J 1999; 13: 1658-1676. [ Links ]
168. Rouquette C, De Chastellier C, Nair S, Berche P. The ClpC ATPase of Listeria monocytogenes is a General Stress Protein Required for Virulence and Promoting Early Bacterial Escape from the Phagosome of Macrophages. Mol Microbiol 1998; 27: 1235-1245. [ Links ]
169. Rouquette C, Ripio M T, Pellegrini E, Bolla J M, Tascon R I, Vázquez-Boland J A, Berche P. Identification of a ClpC ATPase Required for Stress Tolerance and in Vivo Survival of Listeria monocytogenes. Mol Microbiol 1996; 21: 977-987. [ Links ]
170. Samuelsson S, Rothgardt N P, Carvajal A, Fredriksen W. Human Listeriosis in Denmark 1981-1987, Including an Outbreak November 1985-March 1987. J Infectol 1990; 20: 521-259. [ Links ]
171. Santiago N, Zipf A, Bhunia A. Influence of Temperature and Growth Phase on Expression of a 104- kilodalton Listeria Adhesion Protein in L. monocytogenes. App Env Microbiol 1999; 65(6): 2765 - 2769. [ Links ]
172. Schlech W. Overview of Listeriosis. Food Control 1996; 7(4): 183-186. [ Links ]
173. Schubert W D. Internalins from the Human Pathogen Listeria monocytogenes Combine Three Distinct Folds into a Contiguous Internalin Domain. J Mol Biol 2001; 312: 783-794. [ Links ]
174. Schuchat A. Listeriosis and Pregnancy: Food for Thought. Obst Gynecol Surv 1997; 52: 721-722. [ Links ]
175. Schuchat A, Swaminathan B, Broome C V. Epidemiology of Human Listeriosis. Clin Microbiol Rev1991; 4: 169-183. [ Links ]
176. Seeliger H P R, Jones, D. Genus Listeria Pirie. 1986. Bergey’s Manual of Systematic Bacteriology. Sncath P H A, Mair N S, Sharpe M E, Holt J G. Baltimore, Williams and Wilkins. 2: 1235-1245. [ Links ]
177. Sheehan B, Kocks C, Dramsi S, Gouin E, Klarsfeld A D, Cossart P. Molecular and Genetic Determinants of the Listeria monocytogenes Infectious Process. Curr Top Microbiol Inmunol 1994; 192: 187-216. [ Links ]
178. Shen H, Tato C M, Fan X. Listeria monocytogenes as a Probe to Study Cell-Mediated Inmunity. Curr Opi Inmunol 1998; 10: 450-458. [ Links ]
179. Shiloh M U, MacMicking J D, Nicholson S, Brause J E, Potter S, Marino M, Fang F, Dinauer M, Nathan C. Phenotype of Mice and Macrophages Deficient in Both Phagocyte Oxidase and Inducible Nitric Oxide Sythase. Immun 1999; 10: 29-38. [ Links ]
180. Sibelius U, Schulz E C, Rose F, Hattar K, Jacobs T, Weiss S, Chakraborty T, Seeger W, Grimminger F. Role of Listeria monocytogenes Exotoxins Listeriolysin and Phosphatidylinositol-especific Phospholipase C in Activation of Human Neutrophils. Infect Inmun 1999; 67: 1125-1130. [ Links ]
181. Siebers A, Finlay B B. M Cells and the Pathogenesis of Mucosal and Systemic Infections. Trends Microbiol 1996; 4: 22-29. [ Links ]
182. Silver H. Listeriosis During Pregnancy. Obs Ginecol Surv 1998; 53(12): 737-740. [ Links ]
183. Skoble J, Portnoy D, Welch M. Three Regions Within ActA Promote Arp2/3 Complex-mediated Actin Nucleation and Listeria Motility. J Cell Biol 2000; 150: 527-538. [ Links ]
184. Slutsker L, Schuchat A. Listeriosis in Humans. 1999. Listeria, Listeriosis and Food Safety. Ryser E T, Marth E H. New York, Márcel Dekker Inc: 75-95. [ Links ]
185. Smith G A, Marquis H, Jones S, Johnston N C, Portnoy D A, Goldfine H. The Two Distinct Phospholipases C of Listeria monocytogenes Have Overlapping Roles in Escape from a Vacuole and Cell-to-Cell Spread. Infect Inmun 1995; 63: 4231-4237. [ Links ]
186. Sokolovic Z, Fuchs A, Wuenscher M, Goebel W. Synthesis of Species-specific Stress Proteins by Virulent Strains of Listeria monocytogenes. Infect Inmunity 1990; 58: 3582-3587. [ Links ]
187. Spyrou N, Anderson M, Foale R. Listeria Endocarditis: Current Management and Patient Outcome-world Literature Review. Heart 1997; 77: 380-383. [ Links ]
188. Swaminathan B, Rocourt J, Bille J. Listeria. 1995. Manual of Clinical Microbiology. Washington, American Society for Microbiology Press: 341-348. [ Links ]
189. Van Leeuwen H C, O’Hare P. Retargeting of the Mitochondrial Protein p32/gC1Qr to a Cytoplasmic Compartment and the Cell Surface. J Cell Sci 2001; 114: 2115-2123. [ Links ]
190. Vander T M M, Hallevy C, Golzman G, Herishanu Y. Listeria monocytogenes Meningitis in a Patient with Chronic Hepatitis C Infection, Treated by Interferon Alfa and Ribavirin, Case Reports. British Infect Soc 2002; 10: 70. [ Links ]
191. Vasioukhin V. Directed Actin Polymerization is the Driving Force for Epithelial Cell-cell Adhesion. The Cell 2000; 100: 209-219. [ Links ]
192. Vasioukhin V, Fuchs E. Actin Dynamics and Cell-cell Adhesion in Epithelia. Curr Opin Cell Biol 2001; 13: 76-84. [ Links ]
193. Vázquez-Boland J, Domínguez-Bernal G, Gónzalez-Zorn B, Kreft JGoebel W. Pathogenicity Islands and Virulence Evolution in Listeria. Microb Infect 2001a; 3: 571-584. [ Links ]
194. Vázquez-Boland J, Domínguez L, Blanco M, Rocourt J, Fernández-Garayzábal J F, Gutiérrez C B, Tascón R IRodríguez-Ferri E F. Epidemiologic Investigation of a Silage-associated Epizootic of Ovine Listeric Encephalitis, Using a New Listeria-selective Enumeration Medium and Phage Typing. American Journal of Veterinary Research 1992a; 53: 368-371. [ Links ]
195. Vázquez-Boland J, Kuhn M, Berche P, Chakraborty T, Domínguez-Bernal G, Goebel W, González-Zorn B, Wehland J, Kreft J. Listeria Pathogenesis and Molecular Virulence Determinants. Clin Microbiol Rev 2001b; 14(3): 584-640. [ Links ]
196. Vázquez-Boland J A, Gamallo J A, Ripio M T, Domínguez-Bernal G, Lara M, Vega Y, Mainar R C, Suárez M. Listeriosis Animal: Aspectos Epidemiológicos y Diagnósticos, Implicaciones en Salud Pública y Situación en España. Med Vet 1996; 13: 333-344. [ Links ]
197. Vázquez-Boland J A, Kocks C, Dramsi S, Ohayon H, Geoffroy C, Mengaud JCossart P. Nucleotide Sequence of the Lecithinase Operon of Listeria monocytogenes and Possible Role of lecithinase in Cell-to-Cell Spread. Infect Inmun 1992b; 60: 219-230. [ Links ]
198. Vázquez G, De Boland A R. Involvement of Protein Kinase C in the Modulation of 1alpha, 25-dihydroxy-vitamin D3-induced 45Ca2+ Uptake in Rat and Chick Cultured Myoblasts. Biochimica Biophy Acta 1996; 1310: 157-162. [ Links ]
199. Vega Y, Dickneite C, Ripio M T, Bockmann R, González-Zorn B, Novella S, Domínguez-Bernal G, Goebel W, Vázquez-Boland J A. Functional Similarities Between the Listeria monocytogenes Virulence Regulator PrfA and Cyclic AMP Receptor Protein: The PrfA* (Gly145Ser) Mutation Increases Binding Affinity for Targer DNA. J Bacteriol 1998; 180: 6655-6660. [ Links ]
200. Von Both U, Otten S, Darbouche A, Domann E, Chakraborty T. Physical and Genetic Map of the Listeria monocytogenes EGD Serotype 1/2a Chromosome. FEMS Microbiol Lett 1999; 175: 281-289. [ Links ]
201. Wadsworth S J, Goldfine, H. Listeria monocytogenes Phospholipase C-dependent Calcium Signaling Modulates Bacterial Entry Into J774 Macrophage -like Cells. Infect Immun 1999; 67: 1770-1778. [ Links ]
202. Wadsworth S J, Goldfine H. Mobilization of Protein Kinase C in Macrophages Induced by Listeria monocytogenes Affects its Internalization and Escape from the Phagosome. Infect Immun 2002; 70: 4650-4660. [ Links ]
203. Wagner R D, Czuprynski C J. Citokine mRNA Expression in Livers of Mice Infected with Listeria monocytogenes. J Leukocyte Biol 1993; 53: 525-531. [ Links ]
204. Welch M D, Rosenblatt J, Skoble J, Portnoy D A, Mitchison T. Interaction of Human Arp2/3 Complex and the Listeria monocytogenes ActA Protein in Actin Nucleation. Science 1998; 281: 105-108. [ Links ]
205. Wesley I V. Listeriosis in Animals. 1999. Listeria, Listeriosis and Food Safety. Ryser E T, Marth, E.H. New York, Marcel Dekker, Inc: 39-73. [ Links ]
206. Whisstock J C, Lesk A M. SH3 Domains in Prokaryotes. Trends in Biochemistry Science 1999; 24: 132-133. [ Links ]
207. Wiedmann M, Arvik T J, Hurley R J, Boor K J. General Stress Transcription Factor sB and ist Role in Acid Tolerance and Virulence of Listeria monocytogenes. J Bacteriol 1998; 180: 3650-3656. [ Links ]
208. Wiedmann M, Bruce J L, Keating C, Johnson A E, McDonough P L, Batt C A. Ribotypes and Virulence Gene Polymorphisms Suggest Three Distinct Listeria monocytogenes Lineages with Differences in Pathogenic Potential. Infect Inmu 1997; 65(7): 2707-2716. [ Links ]
209. Wuenscher M D, Kohler S, Bubert A, Gerike U, Goebel W. The iap Gene of Listeria monocytogenes is Essential for cell Viability, and its Gene Product, p60, Has Bacteriolytic Activity. J Bacteriol 1993; 175: 3491-3501. [ Links ]


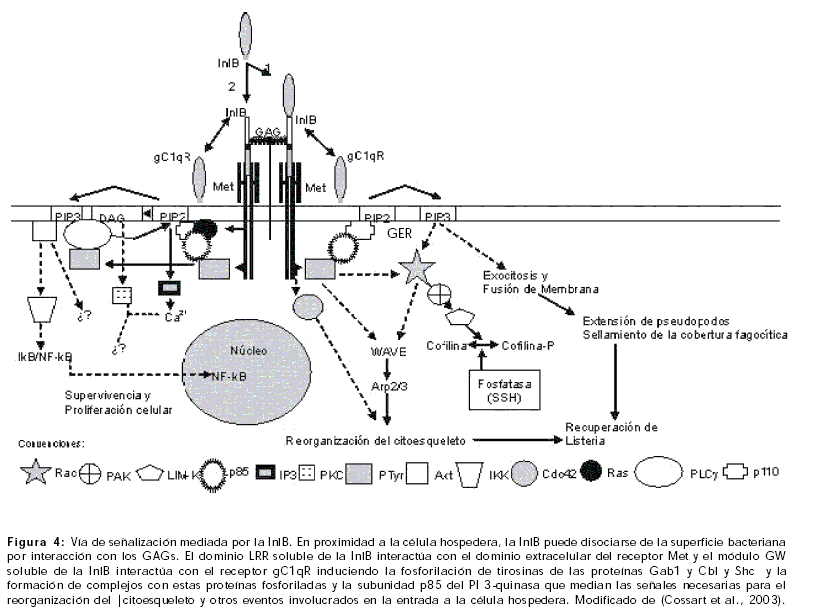{kind=link}