










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkInfectio
Print version ISSN 0123-9392
Infect. vol.15 no.1 Bogotá Jan./Mar. 2011
1 Grupo de Virología, Universidad El Bosque, Bogotá, D.C., Colombia
2 Grupo de Patogénesis Viral, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
Recibido: 9/11/2010; Aceptado: 03/02/2011
Resumen El virus del dengue (DENV) es el agente causal de la enfermedad conocida como dengue, que es la principal enfermedad viral transmitida por artrópodos en el mundo. El DENV es un flavivirus que ingresa por endocitosis y se replica en el citoplasma de la célula infectada, originando tres proteínas estructurales y siete proteínas no estructurales, sobre las cuales se conocen sólo algunas de sus funciones en la replicación viral o en la infección. El ciclo viral que ocurre en las células infectadas hasta ahora está comenzando a aclararse y su conocimiento permitirá en el futuro próximo diseñar racionalmente moléculas que lo intervengan y eviten la replicación del virus. Durante la infección, el individuo puede presentar fiebre indiferenciada o, en otros casos, puede presentar un proceso generalizado de activación de la respuesta inmunitaria innata y adquirida, lo cual provoca la liberación de factores inflamatorios solubles que alteran la fisiología de los tejidos, principalmente el endotelio, conllevando al desarrollo de manifestaciones clínicas graves. Aunque se ha identificado un gran número de factores del individuo asociados al desarrollo de la enfermedad por DENV, queda por identificar el papel de las diferentes proteínas virales en la patogenia de la enfermedad. En la presente revisión, se presenta una breve actualización sobre la estructura y biología del DENV, de su ciclo viral intracelular y, finalmente, se introducen algunos conceptos sobre la inmunopatogenia de la enfermedad producida por este agente.
Palabras clave: virus del dengue, estructura, ensamblaje, inmunopatogenia.
Abstract Dengue virus (DENV) is responsible for the clinical entity known as dengue that is a great concern for economy and public health of tropical countries.
This flavivirus is a single strand RNA virus that after their translation and replication in host cells produces three structural and seven non-structural proteins with specific function in replication or cell binding process that we will describe here. Intracellular viral cycle has begun to be described and this knowledge will impact the rational design of new antiviral drugs. Patients suffering dengue can have an undifferentiated fever or in the severe cases, show an aberrant immunological activation process that lead to soluble inflammatory mediators secretion, affecting tissue function, mainly endothelium. This organ dysfunction is associated with plasma leakage and coagulatory imbalance. Despite it has been recently described some host factors associated with severity of infection, it remains unknown some aspects of viral biology or the role of DENV proteins in disease pathogenesis. This article pretends to make an updated revision about DENV structure, cell viral cycle and introduce some concepts about dengue immunopathogenesis.
Key words: Dengue Virus, structure, assembly, immunopathogenesis
Introducción
El virus del dengue (DENV, acrónimo oficial) pertenece al serocomplejo dengue, género Flavivirus, familia Flaviviridae. Este serocomplejo está conformado por cuatro serotipos denominados DENV1 a DENV4. Los cuatro serotipos circulan periódicamente en áreas endémicas e hiperendémicas y, sin distinción alguna, todos causan la enfermedad conocida como dengue (1).
El DENV es transmitido por mosquitos hembra del género Aedes (especies aegypti y albopictus), distribuidos actualmente en todos los países tropicales y subtropicales del mundo, lo que permite que circulen, cada vez con menos restricciones ecológicas, tanto el virus como el mosquito (2,3). La circulación del DENV entre humanos y mosquitos se presenta cuando el mosquito se alimenta con la sangre de un individuo virémico. Así, el mosquito, al ingerir sangre humana infectada, favorece la infección de las células epiteliales de su intestino; luego, las partículas virales producidas en estas células, son liberadas al hemocele y hacia algunos órganos del mosquito, como las glándulas salivares, las cuales se convierten en órganos reservorios para el virus. La infección en el humano se presenta cuando este mosquito infectado pica nuevamente para alimentarse, liberando saliva y virus.
Luego de cuatro o cinco días, el paciente desarrolla fiebre y dolores generalizados, y se puede detectar virus en la sangre (viremia); después, hay un periodo de disminución de la fiebre y de recuperación que no deja secuelas (4,5). Sin embargo, durante la infección, otros pacientes desarrollan cuadros clínicos más graves, como hemorragias y choque hipovolémico, que pueden dejar secuelas o incluso causar la muerte.
En Colombia, el aumento de casos por dengue en los últimos 10 años ha sido considerable. El Instituto Nacional de Salud reportó para el año 2000 un total de 22.775 casos por dengue, de los cuales, 1.093 fueron dengue grave (manifestaciones más críticas de la enfermedad), con 14 personas fallecidas, mientras que para el año 2009, se reportaron 55.592 casos, de los cuales, 7.131 fueron dengue grave y fallecieron 52 personas.
Para el año 2010, se reportó una epidemia en la que se informaron más de 150.000 casos de dengue, de los cuales, 94% (143.791) fue dengue clásico y el 6% restante correspondió a dengue grave, con una mortalidad de 2,24% (217 casos letales), muy por encima de lo que se reporta normalmente (máximo, 2%).
Los departamentos más afectados por la enfermedad en el 2010 fueron: Antioquia (17,7%), Valle (13,2%), Santander (12,9%), Risaralda (7,7%), Tolima (6,8%) y Quindío (6,0%). Además, durante este periodo se identificaron en circulación los cuatro serotipos de DENV; los más frecuentes fueron DENV1 y DENV2, y la población más afectada por la enfermedad fueron los menores de 15 años (6,7).
Esta situación pone de relieve que en nuestro país el dengue sigue siendo una seria preocupación en salud, pues los factores que agudizan el problema están lejos de solucionarse. Entre estos factores, que hacen previsible la continuidad en el aumento de la morbilidad y mortalidad, se cuentan, por ejemplo, la infestación del mosquito en más de 90% del territorio nacional, el cambio climático y la circulación simultánea de los cuatro serotipos. El aumento en los índices de presencia de mosquitos podría deberse a la resistencia que han venido adquiriendo los vectores al insecticida temefos (8), y al poco impacto que tienen las políticas de prevención y control del vector en las áreas endémicas y en riesgo. Además, el aumento de las poblaciones del vector podría deberse a los cambios en el estilo de vida de las personas, que favorecen la presencia del mosquito en los domicilios que, junto ccon los cambios climáticos, han hecho que los ciclos epidemiológicos sean más cortos. La pobreza extrema y el conflicto armado han obligado al desplazamiento forzado de algunas poblaciones hacia regiones endémicas o con presencia del mosquito, lo cual aumenta las probabilidades de infección y reinfección; esto último podría explicar el aumento de casos por dengue con manifestaciones hemorrágicas (9).
Dada la importancia clínica y epidemiológica que tiene el dengue en nuestro país y en países de Centroamérica y Suramérica, la presente revisión del tema pretende entregar información actualizada sobre el microorganismo en términos de su estructura y ciclo viral, además de algunos elementos generales sobre la inmunopatogenia de la infección por DENV, con el propósito de que sirva como herramienta a los profesionales del sector salud y los profesores y estudiantes, para comprender mejor el reto al cual nos estamos enfrentando.
Virus del dengue
El DENV es un virus icosaedro de 50 nm, aproximadamente, conformado por una membrana lipídica (obtenida de las células del huésped), sobre la cual se insertan las proteínas de membrana y de envoltura. El interior del virus contiene el complejo riboproteico conformado por la proteína de la cápside y el genoma viral que consiste en una única hebra de ARN de sentido positivo que codifica para un polipéptido único, que contiene tanto las proteínas estructurales, que harán parte de la partícula viral, como las proteínas no estructurales, que intervienen durante los procesos de ensamblaje y replicación del ARN genómico, entre otras (figura 1)(1).
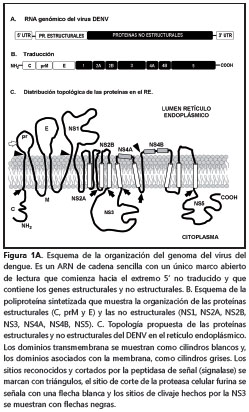
Proteínas virales
Proteínas estructurales. Proteína C. La proteína de la cápside, también conocida como proteína core o de cubierta, pesa 11 kDa, aproximadamente.
Su estructura secundaria consiste en cuatro hélices alfa que cumplen diferentes funciones: las hélices 3 y 4 son hidrofóbicas y anclan la proteína a la membrana del retículo endoplásmico.
La hélice 1, ubicada en el extremo N-terminal de la proteína y orientada hacia el citoplasma, posee aminoácidos de carácter básico que se asocian y unen fuertemente al ARN genómico recién sintetizado; de esta manera, se forma el complejo riboproteico o nucleocápside que protege al ARN viral de la degradación y promueve la organización del ARN en el interior de la partícula viral en formación. La nucleocápside se estabiliza por la interacción de varios homodímeros antiparalelos de la proteína C, que rodean con gran afinidad y especificidad a la hebra de ARN viral (10).
La hélice 2 posee una naturaleza muy hidrofóbica que interviene durante el ensamblaje de la ribonucleoproteína y de la partícula viral. En el primer caso, actúa como una bisagra que favorece el acercamiento del ARN viral al resto de la proteína C anclada en la membrana del retículo endoplásmico. Por otro lado, la hélice 2 recluta pequeñas gotas lipídicas (lipid droplets), presentes en el citoplasma, que promueven la formación de la partícula viral (11). Además, la proteína de la cápside anclada en el retículo endoplásmico interactúa con las proteínas precursora de membrana (prM) y de envoltura, para favorecer y completar el ensamblaje de las partículas virales (1,12,13).
Proteína precursora de membrana (prM) y proteína de membrana (M). La proteína precursora de membrana (prM) tiene un peso molecular de 26 kDa y está presente en los viriones inmaduros y junto con la proteína M, participa fundamentalmente en el proceso de maduración de la partícula viral. La proteína precursora de membrana es procesada después de la transducción por la proteasa celular furina, que la divide en dos y genera, por un lado, el péptido pr, y por otro, la proteína M, que queda con un peso molecular de 8 kDa (1,14). La proteína tiene dos dominios transmembrana y un ectodominio de 40 aminoácidos, aproximadamente (1,15). Este último, según lo descrito por Catteau et al., puede inducir apoptosis en diferentes líneas celulares tumorales. Con el fin de precisar la región del ectodominio que induce apoptosis, mediante técnicas de biología molecular, estos investigadores identificaron un péptido de nueve aminoácidos que corresponde a los residuos 32 al 40 del dominio externo, que fue llamado ApoptoM, como el responsable de inducir la muerte de las células. La señal pro-apoptótica de ApoptoM se induce solamente cuando este dominio es transportado por la ruta secretoria de la célula y se puede inhibir cuando el ectodominio se ancla al retículo endoplásmico o cuando se le adiciona el péptido señal KDEL, que marca a las proteínas para ser devueltas al retículo endoplásmico. Estos resultados sugieren que el péptido ApoptoM de la proteína M podría estar involucrado en la muerte celular y el daño tisular sufrido durante la infección (15).
Proteína de envoltura E. La proteína de envoltura tiene un peso molecular de 50 kDa, posee tres dominios denominados I, II y III, y se distribuye sobre la superficie del virus, formando complejos homodiméricos de tipo cabeza-cola. Los dominios II y III de cada uno de las proteínas del homodímero son determinantes para las interacciones entre el virus y los receptores de las células vulnerables. Por otra parte, la glucoproteína E es el principal inmunógeno del virus, por lo tanto estimula la respuesta inmune del individuo e induce la produccion de anticuerpos neutralizadores.
La importancia funcional de la proteína E radica en que es la única proteína viral que interactúa con las moléculas receptoras de la membrana plasmática de las células vulnerables que favorecen la endocitosis del virus. Por lo tanto, las mutaciones y modificaciones posteriores a la transducción que sufre esta proteína en cada ciclo de replicación, pueden afectar directamente la eficiencia de la replicación, la virulencia y el tropismo del DENV, al igual que puede regular el establecimiento y el control de la infección por parte del sistema inmunitario (4,16-23).
Proteínas no estructurales. NS1, NS2A, NS2B, NS3, NS4A, NS4B y NS5. La función o funciones de cada una de las proteínas no estructurales (NS, non structural proteins) del DENV se han definido parcialmente. A continuación, se describen brevemente algunas de las funciones conocidas de las proteínas no estructurales.
La proteína NS1 (46 kDa) forma dímeros o hexámeros asociados a balsas lipídicas (rafts) de la membrana plasmática (24). También, se puede hallar soluble en el citoplasma y en el espacio extracelular; por esta razón, la NS1 puede estimular al sistema inmunitario.
Varios autores han demostrado en el suero de pacientes infectados con DENV o con el virus de encefalitis japonesa (JVE), la presencia de inmunoglobulinas dirigidas contra la proteína NS1. Estos sueros se han evaluado in vitro y se ha demostrado que las Ig contra la proteína NS1 de ambos virus pueden estimular la lisis mediada por el complemento y dependiente de anticuerpos, tanto en células infectadas como no infectadas.
Este último fenómeno podría explicar, por lo menos en parte, los daños funcionales del endotelio, que conducen al sangrado y a la extravasación plasmática, como se ha demostrado en los pacientes con diagnóstico de dengue grave (21,25-28).
La NS2A es una proteína de 22 kDa, aproximadamente, que in vitro promueve el ensamblaje y la replicación viral. Al parecer, la NS2A coordina de un modo aún no muy bien definido, si el ARN genómico producido en cada ciclo de replicación se utiliza como nueva plantilla para generar las formas replicativas y los intermediarios replicativos o si se asocia dentro de la nucleocápside durante el ensamblaje viral (1). Por su parte, la proteína NS2B (14 kDa) posee una región hidrofóbica que ancla a la membrana del retículo endoplásmico el complejo NS2B/NS3 y luego, por un procesamiento proteolítico, un pequeño dominio hidrofílico de NS2B recién liberado interactúa con el dominio proteasa de la proteína NS3 para actuar como cofactor de ésta (1,29,30).
La proteína NS3 (70 kDa) es una proteína bipartita que posee en el extremo N-terminal un dominio proteasa similar a la tripsina (NS3pro) y en el extremo C-terminal posee un dominio con diferentes actividades enzimáticas, que actúa como trifosfatasa de nucleótidos estimulada por ARN (NTPase) y como helicasa del ARN (NS3Hel); ambas funciones son indispensables en la replicación viral (31,32). El dominio NS3Pro actúa hidrolizando los complejos NS2A/NS2B, NS2B/NS3, NS3/NS4A y NS4B/ NS5 del polipéptido (figura 1).
Como se comentó anteriormente, la función del dominio NS3Pro depende de su asociación con la proteína NS2B, que le confiere estabilidad durante su actividad proteolítica, mientras que la función helicasa permanece inhibida.
Más recientemente, se encontró que la proteína NS3 es la encargada de generar el ambiente lipídico apropiado alrededor del retículo endoplásmico, al reclutar enzimas celulares de la vía de síntesis de lípidos (Fatty Acid Synthase), lo cual garantiza el inicio del ensamblaje (33).
Por otra parte, se ha sugerido que la proteína NS3 puede participar durante los procesos de ensamblaje y de transporte intracelular de los flavivirus. Esta función ha sido sugerida por Patkar et al. (2008), quienes demostraron que la mutación W349A en el dominio helicasa de la proteína NS3 del virus de la fiebre amarilla (YFV) afecta el ensamblaje de las partículas virales sin disminuir la capacidad de replicación del ARN (34).
Por otro lado, Chiou et al, (2003) evidenciaron que la proteína NS3 del JEV se precipita simultáneamente con la proteína TSG101 (Tumor Susceptibility Gene 101), que hace parte del complejo ESCRT I (Endosomal Sorting Complex Required for Transport). Este complejo se forma en el citoplasma y participa en la generación de los cuerpos multivesiculados de la célula, los cuales intervienen en procesos de reciclaje y degradación de proteínas. La proteína TSG101 ha sido reportada como una de las principales proteínas celulares que promueven el ensamblaje del virus de inmunodeficiencia humana-1 (HIV-1) y el virus del Ébola (35). La otra función del la proteína NS3 es actuar como helicasa (NS3Hel), desenrollando las estructuras secundarias que se forman en el extremo 3´ del ARN viral, para favorecer la unión de la polimerasa NS5 sobre el ARN y dar inicio a la replicación (30).
Por último, la proteína NS5 es la más conservada entre todos los flavivirus. Esta proteína es multifuncional, ya que el extremo N-terminal posee actividad enzimática de metiltransferasa y guanidiltransferasa, responsables del capping y la metilación del extremo 5´ del ARN genómico, mientras que, en el extremo C-terminal, se ubica el dominio de ARN polimerasa dependiente de ARN (RdRps) (37). Por lo tanto, la proteína NS5 actúa como la única polimerasa durante la replicación y transcripción virales. Aunque estos procesos suceden exclusivamente en el citoplasma de la célula infectada, se ha identificado una señal de localización nuclear en la proteína NS5 que facilita su importación al núcleo; sin embargo, la razón y la función de la NS5 en el núcleo no se conocen (30,36,38).
Ciclo viral intracelular Entrada, fusión y denudación de la partícula La entrada del virus en células mamíferas y en las de mosquito se inicia con el acercamiento del DENV a la superficie de la célula; luego, la proteína E interactúa con proteínas o proteoglucanos de la membrana celular que median la unión y la posterior endocitosis del virus (14,17,29) (figura 2).
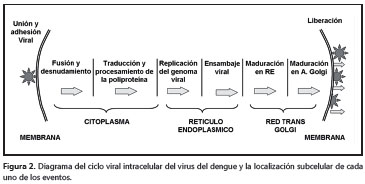
Experimentalmente, se ha demostrado que el dominio III de la proteína E interactúa con el receptor para laminina LAMR1 (39,40), la proteína de adhesión celular ICAM-3 o DC-SIGN (Dendritic Cell- Specific Intercellular Adhesion Molecule-3-Grabbing Non-integrin, CD209) (41) y con proteoglucanos como el heparán sulfato (42), entre otras moléculas. La participación de la proteína DC-SIGN en la adsorción de DENV, fue demostrada por Tassaneetrithep et al. tras transfectar una población de células dendríticas resistentes a la infección, las cuales, cuando expresaron establemente el receptor, se volvieron vulnerables a la infección con los cuatro serotipos de DENV (41).
Tio et al. (2005) sometieron un homogenizado de células de riñón de cerdo (línea PS, clon D) a una electroforesis en dos dimensiones y una transferencia sobre nitrocelulosa; luego, evaluaron con un ensayo de VOPBA (Virus Overlay Protein Binding Assay) las posibles interacciones entre las diferentes proteínas celulares y cada uno de los cuatro serotipos del DENV. Las proteínas que se unieron selectivamente a cada uno de los virus, fueron analizadas por espectrometría de masas de tipo MALDI-TOF, demostrando de esta forma que el receptor para laminina LAMR1 interactúa específicamente con la proteína E de los serotipos 1, 2 y 3 de DENV, lo cual sugiere que esta proteína es un posible receptor viral (39). Estos hallazgos indicarían que, dependiendo del tipo celular, los diferentes serotipos virales pueden utilizar diferentes moléculas receptoras.
Por otro lado, se ha demostrado que, para favorecer la entrada del virus a las células, participan los glucosaminoglucanos o proteoglucanos presentes en la matriz extracelular o que están asociados a las proteínas de superficie de las células. Los proteoglucanos como el heparán sulfato, por su alta carga negativa, pueden actuar como un receptor primario para favorecer el acercamiento de las partículas virales a la superficie celular y, una vez establecido este acercamiento, facilitarían la interacción de la proteína E con proteínas de la superficie para favorecer la endocitocis del virus (42).
El sistema de correceptores es el utilizado por el virus HIV-1 que se une inicialmente al receptor CD4, para luego interactuar con la molécula CCR5 y finalizar el proceso de entrada (43). La participación de un correceptor para la infección por DENV, podría explicar por qué este virus puede infectar diferentes tipos celulares (44), pues este mecanismo le permitiría al virus interactuar inicialmente con el heparán sulfato presente en casi todos los tipos celulares y luego asociarse con un receptor, que promueva la endocitosis.
Este último evento depende de las clatrinas.
Luego, la vesícula endocítica se transforma en un endosoma temprano y posteriormente en un endosoma tardío, el cual se fusiona con un lisosoma que acidifica el pH de la vesícula. El cambio de pH induce los cambios de conformación del dominio II de la proteína E, que favorecen la exposición y el anclaje inmediato del péptido de fusión a la membrana de la vesícula, lo que conlleva finalmente a la liberación de la nucleocápside al citoplasma (45-49).
Replicación del ARN viral
Cuando la nucleocápside se halla libre en el citoplasma, se inician los procesos de traducción y replicación del ARN (13, 50). El ARN genómico viral del DENV es monocatenario de sentido positivo, con un único marco de lectura que traduce un polipéptido completo, el cual es procesado en el retículo endoplásmico por proteasas celulares y la actividad NS3pro, que libera de forma ordenada a las tres proteínas estructurales (C, prM/M y E) y las siete proteínas no estructurales (NS1, NS2A, NS2B. NS3, NS4A, NS4B, NS5) encargadas de la replicación del genoma y el ensamblaje viral (1).
La replicación del ARN viral es un proceso que no está totalmente entendido; sin embargo, in vitro se han detectado tres especies de ARN, denominadas ARN de 20S, 20/28S y 40S, según el valor del coeficiente de sedimentación. Los ARN de 20S conocidos como formas de replicación, no son degradados por las ARNasas y están constituidos por dos cadenas de ARN cada una con polaridad contraria (negativa y positiva). La existencia de las formas de replicación sugiere que estas formas incluyen los intermediarios negativos que actúan como plantilla para la generación de los ARN de sentido positivo. El otro tipo de ARN, los ARN heterogéneos de 20 a 28S, son denominados intermediarios de replicación y corresponden a hebras de ARN de sentido positivo en proceso de elongación.
Por último, los ARN de 40S pueden ser degradados por ARNasas y, al parecer, es el ARN genómico encontrado en los virus ensamblados; por lo tanto, estos ARN pueden ser utilizados para la traducción proteica o para conformar, junto con proteína C la ribonucleoproteína, los nuevos viriones (1).
Durante la traducción, el polipéptido recién sintetizado es acompañado por las proteínas chaperonas BiP, calnexina y calreticulina; luego, cada una de las proteínas virales se organiza en la membrana del retículo endoplásmico y es procesada por proteasas como la furina, la signalasa o la NS3Pro, para finalmente ser modificadas después de la transducción (plegamiento y glucosilación) (33, 50, 51).
Ensamblaje, maduración y liberación del DENV
Los mecanismos que promueven, regulan y coordinan el ensamblaje del virus, no son conocidos completamente. Sin embargo, por microscopía electrónica y criomicroscopía, se ha sugerido que el proceso de ensamblaje de las partículas del DENV sucede en distensiones del retículo endoplásmico denominadas membranas “convolutas” (convolute), donde ocurre de forma simultánea la traducción de la proteína y el ensamblaje del virus (35).
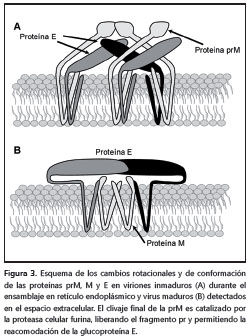
El proceso de ensamblaje comienza con la formación de la nucleocápside gracias a la interacción del ARN genómico y la proteína C en presencia de pequeñas gotas de lípidos (11); sobre esta primera estructura luego se asocian las proteínas prM/M y E, que deben estar inmersas en la membrana del retículo endoplásmico. Posteriormente, suceden dos etapas de maduración de la partícula viral. Primero se organizan de forma heterodimérica las proteínas prM/M y E, en donde la primera recubre a la segunda; este recubrimiento le confiere un aspecto rugoso a la superficie del virus cuando se observa por microscopía electrónica. En el segundo paso, esta partícula inmadura transita desde el retículo endoplásmico hasta las regiones cis y trans del aparato de Golgi, donde se inicia la segunda etapa de maduración. En esta última etapa, los cambios de conformación y de rotación de la proteína E generan homotrímeros antiparalelos de la misma, lo que le da una apariencia lisa a la superficie del virus. Por último, un nuevo procesamiento proteolítico sobre la proteína prM/M por la proteasa furina, independiza el péptido pr y la proteína M. Esta nueva modificación estabiliza los homotrímeros de E y mantiene unido al péptido pr.
Finalmente, cuando el virus es liberado, el pH neutro del espacio extracitoplásmico induce el desprendimiento del péptido pr y la proteína E adquiere la conformación final que puede ser reconocida por las moléculas receptoras de la célula sensible e iniciar un nuevo ciclo de infección en otra célula (figura 3) (13,50,52-57).
Patogenia del dengue
La morbilidad y mortalidad causadas por la infección por DENV, están dadas por la complejidad de eventos que se presentan en el transcurso de la infección. Algunos pacientes desarrollan cuadros febriles y dolores generalizados que se resuelven rápidamente sin dejar secuelas. A este tipo de manifestación clínica se le conoce como dengue (fiebre de dengue). Otros pacientes, por el contrario, presentan dolores intensos, fiebre alta e incrementos en la permeabilidad vascular, lo que conlleva a la pérdida de plasma y hemorragias pleurales y gastrointestinales, entre otros. Estos signos son agrupados en dos entidades clínicas conocidas como dengue con signos de alarma y dengue grave con manifestaciones hemorrágicas o sin ellas, llamado anteriormente dengue hemorrágico, o fiebre hemorrágica por dengue (2,21,58-63).
Las principales células diana de la infección por DENV son los monocitos, los macrófagos, las células dendríticas y los linfocitos CD4+ y CD8+. In vitro se ha reportado que se infectan células del endotelio, varias líneas celulares hepáticas, fibroblásticas y neuronales (44). Una vez establecida la infección en el huésped, las células expresan como primera línea de defensa el interferón (IFN) de tipo I (α y β), que busca inhibir la replicación viral. Por otro lado, se inicia el proceso de presentación de antígenos mediante el complejo mayor de histocompatibilidad (CMH) de tipo I y II, lo que conlleva a que células como las NK (natural killer) ataquen a las células infectadas y liberen, junto a los linfocitos T, el IFN de tipo II (γ). Esta actividad es el fenómeno responsable del control de la infección, ya que se establece un estado antiviral mediado por IFN que evita la replicación del virus en las células infectadas o la infección de nuevas células. Además, esta señalización puede inducir la apoptosis de las células infectadas o alteradas (60).
Por otro lado, los linfocitos T desempeñan un papel preponderante en el establecimiento y control de la respuesta inmunitaria frente al virus. Tanto los linfocitos CD4+ como los CD8+ estimulados por diferentes citocinas, como el IFN (tipo I y II) y el factor de necrosis tumoral alfa (TNFα), se activan y secretan citocinas que pueden tener un carácter proinflamatorio o antiinflamatorio.
En resumen, esta respuesta inmunitaria es la que normalmente se presenta en los pacientes infectados por primera vez que logran resolver la infección; sin embargo, en los pacientes que sufren una nueva infección con un serotipo diferente al que causó la primera (frecuente en zonas endémicas donde circula más de un serotipo de DENV), ocurre un fenómeno que estimula y exacerba la respuesta inmunitaria del paciente, lo que aumenta las probabilidades de que desarrolle dengue grave, con manifestaciones hemorrágicas o sin ellas (5,64-68).
El desarrollo del dengue grave y su asociación con las reinfecciones está bien argumentada clínica y experimentalmente. Una de las teorías más aceptadas y polémica, se denomina potenciación de la infección dependiente o mediada por anticuerpos, que se presenta cuando los anticuerpos producidos y dirigidos contra el serotipo de DENV que causó la infección por primera vez, reconocen y forman complejos con el segundo serotipo de virus causante de la reinfección. Estos complejos virus-anticuerpos se unen a los monocitos y macrófagos mediante los receptores Fc, favoreciendo la penetración del virus. Este mecanismo incrementa la proporción de células infectadas, la viremia y la capacidad de dispersión del virus en el organismo. Esto explica por qué algunos pacientes con dengue grave poseen títulos virales más altos en comparación con los pacientes con dengue sin signos de alarma. Además, el fenómeno de la potenciación de la infección dependiente o mediada por anticuerpos estimula la activación en células como linfocitos y macrófagos, induciendo la liberación de citocinas y otros factores solubles que alteran, entre otros aspectos, la fisiología del tejido endotelial, lo que facilita la extravasación y la formación de edemas, petequias y hemorragias (63,66,69).
La evidencia clínica de la potenciación de la infección dependiente o mediada por anticuerpos está dada por los cientos de casos de dengue grave con manifestaciones hemorrágicas que se han descrito en Tailandia, donde el dengue es hiperendémico y la población más vulnerable a la infección son los menores de 15 años quienes han sufrido por lo menos una infección por DENV (70). Por otra parte, los menores de un año que presentan signos de dengue grave al ser infectados por primera vez por un serotipo de DENV, desarrollan estos signos por la presencia de anticuerpos anti-DENV transmitidos verticalmente por sus madres (71). Sin embargo, la literatura también reporta casos de dengue grave con manifestaciones hemorrágicas en pacientes infectados por primera vez. Esto sugiere que el desarrollo de estas manifestaciones puede tener otras causas adicionales, como la edad de los pacientes (72, 73), el sexo (62) y factores genéticos del individuo, como la raza y algunos polimorfismos asociados a los genes HLA, TNF-α, y CD209 (74-76). Por otra parte, el serotipo y el genotipo del virus también pueden estar relacionados con la gravedad de la enfermedad (2).
Otro mecanismo que se ha asociado al desarrollo de dengue grave, es la lisis de las células endoteliales, mediada por complemento y dependiente de anticuerpos, especialmente aquellos dirigidos contra NS1, que reconocen un antígeno aún no identificado presente en la superficie del endotelio. Esta interacción activa el sistema de complemento que altera la permeabilidad vascular, induce la disfunción del tejido y lisis de las células endoteliales (24,25,64,77,78).
Por otro lado, la gravedad de la enfermedad puede deberse a las grandes concentraciones y a la constante permanencia de algunas de citocinas producidas y liberadas por células como linfocitos, macrófagos y endoteliales, entre otras. Como se dijo anteriormente, las células infectadas y las no infectadas, responden al estímulo inducido por IFN de tipo I y II que activan sobre éstas, la proliferación, la diferenciación y la apoptosis. Además, pueden estimular la expresión de algunas moléculas de adhesión y de receptores que promueven de nuevo la expresión de citocinas y otros mediadores solubles (61).
Esta activación inmunitaria constante sostiene una señalización que afecta a las células, alterando la función del endotelio, de los linfocitos y de los macrófagos (60,61,79). Entre los mediadores solubles que se han detectado en pacientes infectados con DENV, se encuentran citocinas de tipo Th1 y Th2 (T helper lymphocytes) secretadas por linfocitos CD4+ o CD8+ (5,60,61,79,80). En pacientes con diagnóstico de dengue sin signos de alarma, se detectan citocinas de tipo Th1 como IFNγ e interleucina 2 (IL-2), mientras que, en los pacientes con dengue grave, se detectan citocinas de tipo Th2, como las IL-4, IL-6, IL-8 e IL-10. Particularmente, la IL-8 se presenta en grandes concentraciones en el suero de estos pacientes y, en algunos casos, este incremento se asocia con un aumento en la permeabilidad vascular, la efusión pleural y la muerte de los pacientes (68).
El otro grupo de moléculas que se expresa en exceso en las células infectadas y no infectadas, son las moléculas de adhesión como ICAM-1, VCAM-1, E, L y P-selectina, entre otras. Estas moléculas facilitan el reconocimiento, la unión y la posterior diapédesis de células como los monocitos, que atraviesan la barrera endotelial y circulan en los espacios intersticiales. Esto permite, por un lado, la propagación del virus a otras células y tejidos, y, por otro, el paso de líquido y mediadores solubles que estimulan los procesos inflamatorios (5, 66, 78).
En resumen, durante la infección por DENV la respuesta inmunitaria puede resolver la infección, sin causar grandes traumatismos en el individuo o, por el contrario, puede llevar al organismo a un aparente caos, donde la constante estimulación conlleva a la activación celular, el aumento de la expresión de mediadores y de receptores que inducen en algunos casos daños tisulares irreversibles, lo que aumenta la gravedad de la enfermedad (4, 46, 60, 61, 63, 74).
Finalmente, aunque en los últimos años se han definido e identificado varios factores que pueden favorecer directa o indirectamente el desarrollo de las formas más graves de dengue, no se han establecido con claridad las principales causas que incrementan de forma notoria la respuesta inmunitaria en algunos pacientes. Además, el aumento de casos de dengue con manifestaciones atípicas, como miocarditis, encefalitis, hepatitis o insuficiencia renal, sugiere cambios en el perfil de la enfermedad que podrían deberse a cambios del tropismo del virus; esto último muestra la necesidad de conocer más sobre el virus y los posibles mecanismos que está utilizando para infectar diferentes tipos celulares o diferentes tejidos.
En los últimos años, a partir del conocimiento de la estructura del virus, se ha propuesto, por ejemplo, el bloqueo de la actividad NS3pro con inhibidores específicos, el bloqueo de la polimerasa NS5 con nucleótidos modificados y, más recientemente, se está usando la detección de la proteína NS1 en el suero, como una prueba diagnóstica de mayor sensibilidad.
Es indudable que el nuevo conocimiento sobre la estructura y la función de las diferentes proteínas virales y sobre el ciclo viral, aumenta las posibilidades de plantear nuevas estrategias farmacológicas o vacunales que permitan evitar o tratar la enfermedad.
Agradecimientos Esta revisión fue hecha en el marco del proyecto “Neuroinmunología de la infección in vivo por el virus del dengue” financiado por del Departamento Administrativo de Ciencia, Tecnología e Innovación, Colciencias (código 130-80517588), la Universidad El Bosque y la Facultad de Odontología de la Universidad Nacional de Colombia. Myriam L. Velandia fue becaria del Programa de Formación de Doctorados de Colciencias.
Correspondencia: Jaime E. Castellanos, Carrera 30 Nº 45-03, Facultad de Odontología, Edificio 210, Oficina 210, Bogotá, D.C., Colombia. Teléfono: (571) 316-5555; fax: (571) 648-9066. Dirección electrónica: jecastellanosp@unal.edu.co
Referencias
1. Lindenbach B, Thiel H, Rice C. Flavivirus: The virus and their replication. In: Knipe D, Howley Peter. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 1101-52. [ Links ]
2. Kyle J, Harris E. Global spread and persistence of dengue. Annual Rev Microbiol. 2008;62:71-92. [ Links ]
3. Tolle M. Mosquito-borne diseases. Curr Probl Pediatr Adolesc Health Care. 2009;39:97-140. [ Links ]
4. McBride W, Bielefeldt H. Dengue viral infections; pathogenesis and epidemiology. Microb Infect. 2000;2:1041-50. [ Links ]
5. Lei H, Yeh T, Liu H, Lin Y, Chen S, Liu C. Immunopathogenesis of dengue virus infection. J Biomed Sci. 2001;8:377-88. [ Links ]
6. Ministerio de la Protección Social, Instituto Nacional de Salud. Subdirección de Vigilancia y Control en Salud Pública. Inf Quinc Epidemiol Nac. 2010;15:381-4. [ Links ]
7. Instituto Nacional de Salud, Subdirección de Vigilancia y Control en Salud Pública. Boletín Epidemiológico Semanal. Semana epidemiológica 51 de 2010 (19 al 25 de diciembre de 2010). Internet. Diciembre de 2010. Disponible en: http://190.27.195.165:8080/index.php?idcategoria=83087# [ Links ]
8. Maestre R, Rey G, De Las Salas A, Vergara C, Santacoloma L, Goenaga S, et al. Susceptibilidad de Aedes aegypti (Diptera: Culicidae) a temefos en Atlántico-Colombia. Rev Colomb Entomol. 2009;35:202-5. [ Links ]
9. Ministerio de la Protección Social, Instituto Nacional de Salud, OPS/ OMS. Guía de atención clínica integral del paciente con dengue. Bogotá: Ministerio de la Protección Social; 2010. [ Links ]
10. Schneemann A. The structural and functional role of RNA in icosahedral virus assembly. Annu Rev Microbiol. 2006;60:51-67. [ Links ]
11. Samsa M, Mondotte J, Iglesias N, Assunção I, Barbosa G, Da Poian A, et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2005;10:e1000632. [ Links ]
12. Ma L, Jones C, Groesch T, Kuhn R, Post C. Solution structure of dengue virus capsid protein reveals another fold. Proc Natl Acad Sci USA. 2004;101:3414-9. [ Links ]
13. Urbanowski M, Ilkow C, Hobman T. Modulation of signaling pathways by RNA virus capsid proteins. Cell Signal. 2008;20:227-36. [ Links ]
14. Zhang Y, Cover J, Chipman P, Zhang W, Pletnev S, Sedlak D, et al. Structures of immature flavivirus particles. EMBO J. 2003;22:2604-13. [ Links ]
15. Catteau A, Kalinina O, Wagner M, Deubel V, Courageot M, Despres P. Dengue virus M protein contains a proapoptotic sequence referred to as ApoptoM. J Gen Virol. 2003;84:2781-93. [ Links ]
16. Imbert J, Guevara P, Ramos-Castañeda J, Ramos C, Sotelo J. Dengue virus infects mouse cultured neurons but not astrocytes. J Med Virol. 1994;42:228-33. [ Links ]
17. Allison S, Schalich J, Stiasny K, Mandl C, Kunz C, Heinz F. Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acid pH. J Virol. 1995;69:695-700. [ Links ]
18. Chem Y, Maguire T, Hileman R, Fromm J, Esko J, Linhadt R, et al. Dengue virus infectivity depends on envelope protein binding to target cell heparin sulfate. Nat Med. 1997;3:866-71. [ Links ]
19. Hung S, Lee P, Chen H, Chen L, Kao C, King C. Analysis of the steps involved in dengue virus entry into host cells. Virology. 1999;257:156-67. [ Links ]
20. Lee E, Lobigs M. Substitution at the putative receptor-binding site of an encephalitic flavivirus alter virulence and host cell tropism and reveal a role for glycosaminoglycans in entry. J Virol. 2000;74:8867-75. [ Links ]
21. Lin C, Lei H, Shiau A, Liu H, Yeh T, Chen S, et al. Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J Immunol. 2002;169:657-64. [ Links ]
22. Zhang Y, Zhang W, Ogata S, Clements D, Strauss J, Baker T, et al. Conformational changes of the flavivirus E glycoprotein. Structure. 2004;12:1607-18. [ Links ]
23. Stiasny K, Heinz F. Flavivirus membrane fusion. J Gen Virol. 2006;87:2755-66. [ Links ]
24. Noisakran S, Dechtawewat T, Avirutnan P, Kinoshita T, Siripanyaphinyo U, Puttikhunt C, et al, Association of dengue virus NS1 protein with lipid rafts. J Gen Virol. 2008;89:2492-500. [ Links ]
25. Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, et al. Vascular leakage in severe dengue virus infections: A potential role for the nonstructural viral protein NS1 and complement. J Infect Dis. 2006;193:1078-88. [ Links ]
26. Avirutnan P, Zhang L, Punyadee N, Manuyakorn A, Pittikhunt C, Kasinrerk W, et al. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 2007;3:e183. [ Links ]
27. Schelesinger J. Flavivirus nonstructural protein NS1: Complementary surprises. Proc Natl Acad Sci USA. 2006;103:18879-80. [ Links ]
28. Krishna VD, Rangappa M, Satchidanandam V. Virus-specific cytolytic antibodies to nonstructural protein 1 of Japanese encephalitis virus effect reduction of virus output from infected cells. J Virol. 2009;83:4766-77. [ Links ]
29. Lindenbach B, Rice C. Molecular biology of flavivirus. Adv Virus Res.2003;59:23-61. [ Links ]
30. Bollati M, Álvarez K, Assenberg R, Baronti C, Canard B, Cook S, et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antiviral Res. 2010;87:125-48. [ Links ]
31. Bazan J, Fletterick R, Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology. 1989;171:637-9. [ Links ]
32. Chambers T, Weir R, Grakoui A, McCourt D, Bazan J, Fletterick R, et al. Evidence that the N-terminal domain of nonstructuralprotein NS3 from yellow fever virus is a serine protease responsible for sitespecific cleavages in the viral polyprotein. Proc Natl Acad Sci USA. 1990;87:8898-02. [ Links ]
33. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A. 2010;107:17345-50. [ Links ]
34. Patkar C, Kuhn R. Yellow Fever Virus NS3 plays and essential role in virus assembly independent of its enzymatic functions. J Virol. 2008;82:3342-52. [ Links ]
35. Chiou C, Andrew C, Chen P, Liao C, Lin Y, Wang J. Association of Japanese encephalitis virus NS3 protein with microtubules and tumor susceptibility gene 101 (TSG101) protein. J Gen Virol. 2003;84:2795-05. [ Links ]
36. Muñoz-Jordan JL, Sánchez-Burgos GG, Laurent-Rolle M, García- Sastre A. Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci USA. 2003;100:14333-8. [ Links ]
37. Selisko B, Peyrane FF, Canard B, Álvarez K, Decroly E. Biochemical characterization of the (nucleoside-2âºO)-methyltransferase activity of dengue virus protein NS5 using purified capped RNA oligonucleotides (7Me) GpppAC(n) and GpppAC(n). J Gen Virol. 2010;91:112-21. [ Links ]
38. Nomaguchi M, Ackermann M, Yon C, You S, Padmanabhan R. De novo synthesis of negative-strand RNA by dengue virus RNA-dependent RNA polymerase in vitro: Nucleotide, primer and template parameters. J Virol. 2003;77:8831-42. [ Links ]
39. Tio P, Jong, W, Cardosa, M. Two dimensional VOPBA reveals laminin receptor (LAMR1) interaction with dengue virus serotypes 1, 2 and 3. Virol J. 2005;2:25-36. [ Links ]
40. Nelson J, McFerran N, Pivato G, Chambers E, Doherty C, Steele D, et al. The 67 kDa laminin receptor: Structure, function and role in disease. Biosci Rep. 2008;28:33-48. [ Links ]
41. Tassaneetrithep B, Burgess T, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197:823-9. [ Links ]
42. Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW, et al. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology. 2002;292:162-8. [ Links ]
43. Mosier D. How HIV changes its tropism: Evolution and adaptation?. Curr Opin HIV AIDS. 2009;4:125-30. [ Links ]
44. Diamond MS, Edgil D, Roberts TG, Lu B, Harris E. Infection of human cells by dengue virus is modulated by different cell types and viral strains. J Virol. 2000;74:7814-23. [ Links ]
45. Mosso C, Galván-Mendoza I, Ludert J, Ángel R. Endocytic pathway followed by dengue virus to infect the mosquito cell line C6/36 HT. Virology. 2009;378:193-9. [ Links ]
46. Rothman A, Ennis F. Toga/Flaviviruses: Immunophathology. In: Cunnigham M, Fujinami R, editors. Effects of microbes on the immune system. Philadelphia PA: Lippincott Williams & Wilkins; 2000. p. 473-90. [ Links ]
47. van der Schaar H, Rust M, Waarts B, van der Ende-Metselaar H, Kuhn R, Wilschut J, et al. Characterization of the early events in dengue virus cell entry by biochemical assay and single-virus tracking. J Virol. 2007;81:12019-28. [ Links ]
48. van der Schaar H, Rust M, Chen C, van der Ende-Merselaar H, Wilschur J, Zhuang X, et al. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008; 4:e1000244. [ Links ]
49. van der Schaar H, Wilschut J, Smit J. Role of antibodies in controlling dengue virus infection. Inmunobiology. 2009;214:613-29. [ Links ]
50. Qi R, Zhang L, Chi C. Biological characteristics of dengue virus and potential targets for drug design. Acta Biochim Biophys Sin. 2008;40:91-101. [ Links ]
51. Limjindaporn T, Wongwiwat W, Noisakran S, Srisawat C, Netsawang J, Puttikhunt C, et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem Biophys Res Commun. 2009;379:196-200. [ Links ]
52. Elsuhuber S, Allison SL, Heinz F, Mandl C. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J Gen Virol. 2003;84:183-91. [ Links ]
53. Jones C, Ma L, Burgener J, Groesch T, Post C, Kuhn R. Flavivirus capside protein is a dimeric alpha-helical protein. J Virol. 2003;77:7143-9. [ Links ]
54. Kiermayr S, Kofler R, Mandl C, Messner P, Heinz F. Isolation of capsid protein dimers from the tick-borne encephalitis flavivirus and in vitro assembly of capsid-like particles. J Virol. 2004;78:8078-84. [ Links ]
55. Wang S, Syu W, Hu S. Identification of the homotypic interaction domain of the core protein of dengue virus type 2. J Gen Virol. 2004;85:2307-14. [ Links ]
56. Perera R, Khaliq M, Kuhn R. Closing the door on flaviviruses: Entry as a target for antiviral drug design. Antiviral Res. 2008;80:11-22. [ Links ]
57. Perera R, Kuhn R. Structural proteomics of dengue virus. Curr Opin Microbiol. 2008;11:369-77. [ Links ]
58. Gubler D. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480-96. [ Links ]
59. Rothman A. Dengue: Defining protective versus pathologic immunity. J Clin Invest. 2004;113:946-51. [ Links ]
60. Pang T, Cardosa M, Guzmán M. Of cascades and perfect storms: The immunopathogenesis of dengue haemorrhagic fever-dengue shock syndrome (DHF/DSS). Immunol Cell Biol. 2007;85:42-5. [ Links ]
61. King N, Shrestha B, Kesson A. Immune modulation by flaviviruses. Adv Virus Res. 2003;60:121-55. [ Links ]
62. Thomas S, Strickman D, Vaughn D. Dengue epidemiology: Virus epidemiology, ecology and emergence. Adv Virus Res. 2003;61:235-89. [ Links ]
63. Oishi K, Saito M, Mapua C, Natividad F. Dengue illness: Clinical features and pathogenesis. J Infect Chemother. 2007;13:125-33. [ Links ]
64. Lin C, Wan S, Cheng H, Lei H, Lin Y. Autoimmune pathogenesis in dengue virus infection. Viral Immunol. 2006;19:127-32. [ Links ]
65. Martínez-Gutiérrez M, Castellanos JE. Dengue hemorrágico, ¿una aberración inmunológica? Revista Escuela Colombiana de Medicina. 2006;11:10-9. [ Links ]
66. Kou Z, Quinn M, Chen H, Rodrigo W, Rose R, Schlesinger J, et al. Monocytes but not T or B cells, are the principal target cells for dengue virus (DV) infection among human peripheral blood mononuclear cells. J Med Virol. 2008;80:134:46. [ Links ]
67. Halstead S. Dengue. In: Pasvol G, Hoffman S. Tropical medicine science and practice. Singapore: Imperial College Press; 2008. p. 285-326. [ Links ]
68. Srikiatkhachorn A. Plasma leakage in dengue hemorrhagic fever. Thromb Haemost. 2009;102:1042-9. [ Links ]
69. Guzmán M. Deciphering dengue: The cuban experience. Science. 2005;309:1495-7. [ Links ]
70. Murge B. Severe dengue: Questioning the paradigm. Microb Infect. 2010;12:113-8. [ Links ]
71. Martina B, Koraka P, Osterhaus A. Dengue virus pathogenesis: An integrated view. Clin Microbiol Rev. 2009;22:564-81. [ Links ]
72. Kabra S, Jain Y, Pandey R, Singhal M, Tripathi P, Seth B , et al. Dengue haemorrhagic fever in children in the 1996 Delhi epidemic. Trans R Soc Trop Med Hyg. 1999;93:294-8. [ Links ]
73. Hongsiriwon S. Dengue hemorrhagic fever in infants. Southeast Asian J Trop Med Public Health. 2002;33:49-55. [ Links ]
74. Fernández-Mestre MT, Gendzekhadze K, Rivas-Vetencourt P, Layrisse Z. TNFalpha 308-A allele, a possible severity risk factor of hemorrhagic manifestation in dengue fever patients. Tissue Antigens. 2004;64:469-72. [ Links ]
75. Chaturvedi U, Nagar R, Shrivastava R. Dengue and dengue hemorrhagic fever: Implications of host genetics. FEMS Immunol Med Microbiol. 2006;47:155-66. [ Links ]
76. Chao Y, Huang C, Lee C, Chang S, King C, Kao C. Higher infection of dengue virus serotype 2 in human monocytes of patients with G6PD deficiency. Plos ONE. 2008;3:e1557. [ Links ]
77. Warke R, Xhaja K, Martin K, Fournier M, Shaw S, Brizuela N, et al. Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J Virol. 2003;77:11822-32. [ Links ]
78. Green S, Rothman A. Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr Opin Infect Dis. 2006;19:429-36. [ Links ]
79. Chaturvedi U, Agarwal R, Elbishbish E, Mustafa A. Cytokine cascade in dengue hemorrhagic fever: Implications for pathogenesis. FEMS Immunol Med Microbiol. 2000;28:183-8. [ Links ]
80. Houghton-Triviño N, Salgado D, Rodríguez J, Bosch I, Castellanos JE. Levels of soluble ST2 in serum associated with severity of dengue due to tumor necrosis factor alpha stimulation. J Gen Virol. 2010; 91:697-06. [ Links ]

