










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkDYNA
versão impressa ISSN 0012-7353versão On-line ISSN 2346-2183
Dyna rev.fac.nac.minas v.75 n.154 Medellín jan./mar. 2008
TOLERANCIA AL CO EN CELDAS DE COMBUSTIBLE
CO TOLERANCE IN FUEL CELLS
BIBIAN HOYOS
Escuela de Procesos y Energía, Universidad Nacional de Colombia, Sede Medellín, bahoyos@unal.edu.co
NATALIA MUNERA
Universidad Nacional de Colombia, Facultad de Minas, sede Medellín
FARID CHEJNE
Escuela de Procesos y Energía, Universidad Nacional de Colombia, Sede Medellín, fchejne@unal.edu.co
Recibido para revisar Abril 18 de 2007, aceptado Noviembre 30 de 2007, versión final Diciembre 12 de 2007
RESUMEN: El entendimiento completo del proceso de adsorción y posterior oxidación de moléculas de CO en platino es de fundamental importancia para el desarrollo de celdas de combustible poliméricas que operan a baja temperatura. En este trabajo se presenta una revisión de las cinco estrategias experimentales más importantes en la búsqueda de mejorar la tolerancia al CO: disminución del potencial de inicio de la reacción de oxidación, reducción de la cantidad de CO adsorbido, utilización de pequeñas cantidades de oxígeno en la corriente de alimentación al ánodo, aumento de la temperatura de operación y limpieza del CO a la entrada.
Aunque se han desarrollado catalizadores bastante promisorios (PtMo y PdAu), todavía se sigue considerando a la mezcla Pt-Ru como el catalizador anódico más eficiente para combustibles que contienen 10 ppm de CO o más.
La estrategia de inyectar oxígeno al ánodo parece promisoria pero requiere el desarrollo de nuevas membranas más resistentes y de la implementación de condiciones más seguras de operación de la celda.
El diseño estructural de ánodos especiales con múltiples capas soportando catalizadores específicos para cada tipo de combustible puede ser una estrategia muy atractiva.
PALABRAS CLAVE: Tolerancia al CO, Celdas de combustible. Catalizadores basados en platino.
ABSTRACT: Full comprehension of adsorption and further oxidation of CO molecules on platinum is fundamental to develop polymer exchange membrane fuel cells operating at low temperature. In this work it is presented an overview of the five more important experimental strategies in the search to improve the CO tolerance: reduce the on set potential for the CO oxidation reaction, reduce the amount of adsorbed CO, introduce small quantities of oxidant bleeding into the anode feed, increase the operation temperature and cleaning the CO in the fuel feed.
Even though quite promissory catalysts have been developed (PtMo and PdAu mixtures), still the PtRu systems are regarded as the most efficient anode catalysts for fuels containing over 10 ppm of CO.
The strategy of inject oxygen to the anode appears promissory, but it requires the development of new more efficient membranes and the implementation of more safe operation conditions of the fuel cell.
The structural design of special anodes containing multiple layers with specific catalysts for each type of fuel can become a very attractive strategy.
KEYWORDS: Carbon monoxide tolerance; Proton-exchange membrane fuel cell, Platinum-based catalyst.
1. INTRODUCCIÓN
Las celdas de combustible son dispositivos que permiten la conversión directa de energía química en electricidad sin la necesidad de etapas de combustión intermedias. Debido a que dentro del dispositivo no se tienen componentes móviles, las pérdidas ocasionadas por fricción e irreversibilidades disminuyen notablemente, comparadas con las pérdidas experimentadas en un motor de combustión interna, razón por la cual una celda de combustible puede alcanzar eficiencias considerablemente altas.
Actualmente, las aplicaciones vehiculares son las que atraen mayor interés, debido a la demanda creciente de automóviles con motores más eficientes y menos perjudiciales para el medio ambiente. Sin embargo, las dificultades técnico-económicas para obtener hidrógeno de alta pureza, ponen en evidencia que para la introducción masiva y la popularización de vehículos eléctricos que operen con celdas de combustible de baja temperatura, se vislumbran dos posibles escenarios a corto plazo: El primero es producir en el mismo vehículo una corriente rica en hidrógeno a partir de hidrocarburos o alcoholes mediante procesos de reformado con vapor de agua. El segundo escenario es la alimentación directa del combustible a la celda.
En cualquiera de los dos casos, la existencia de CO en el ánodo de la celda constituye un inconveniente en el desarrollo y posterior funcionamiento del dispositivo: hasta ahora, el platino es considerado el mejor catalizador para la oxidación de hidrógeno puro, y presenta las mejores características de adsorción de combustibles que tienen átomos de carbono (hidrocarburos, alcoholes, etc.). El principal problema para el desarrollo de celdas de combustible alimentadas con estos combustibles es la rápida pérdida de actividad (envenenamiento) del platino utilizado en el ánodo, debido a una adsorción fuerte de CO sobre la superficie, lo que inhabilita los sitios activos para una posterior reducción del combustible. Este hecho ha conducido a la generación de una gran cantidad de investigación experimental para encontrar un catalizador apropiado para la oxidación total de CO (Gaisteger et al., 1995a,b,c; Couto et al., 1996; Grgur et al., 1998; Jusys et al., 1999). En esta búsqueda se han ensayado muchas formulaciones de catalizadores para intentar mejorar la velocidad de oxidación.
El envenenamiento de la superficie de platino con CO lleva a un decrecimiento significativo en la potencia de salida de la celda. Por ejemplo, cantidades tan pequeñas como 100 ppm de CO en el gas del ánodo reduce la potencia de salida de la celda a sólo el 25% del valor que se obtiene utilizando hidrógeno puro. Recientemente, Papageorgopoulos y de Bruijn (2002) investigaron cuantitativamente el envenenamiento de platino expuesto a una mezcla de 1% CO / H2 y encontraron que a 25 °C, el CO bloquea el 98% de los sitios activos.
Gottesfeld y Pafford (1998) mostraron que el desempeño de las celdas de combustible es altamente dependiente de la concentración de CO alimentado. Adicionalmente, un trabajo experimental bastante detallado sobre el efecto de la concentración de CO en el desempeño de celdas de combustible poliméricas fue reportado por Oetjen et al. (1996). Dichos estudios muestran que inclusive 5 ppm de CO en la corriente de hidrógeno puede causar una disminución en el desempeño para densidades de corriente superiores a 400 mA/cm2. Las pérdidas de voltaje debidas al envenenamiento con CO son más dramáticas inicialmente con el incremento en la concentración de CO en el ánodo. Es decir, a una densidad de corriente de 700 mA/cm2 la pérdida de voltaje es alrededor de 400 mV cuando la concentración de CO se incremente de 0 a 20 ppm, mientras que la subsiguiente pérdida de voltaje es de 100 mV para aumentos en la concentración de CO de 20 a 100 ppm.
En la literatura de los últimos años concerniente a la adsorción y oxidación de CO en catalizadores anódicos para celdas de combustible se pueden observar dos tendencias claras: un gran número de publicaciones (Brako y Sokcevic, 2001; Christensen et al., 2004; Gaisteger et al., 2004; Spendelow et al., 2004) que contienen la búsqueda de datos experimentales en los cuales se quiere lograr un entendimiento básico de los mecanismos de adsorción y oxidación de CO en diferentes catalizadores, con un gran énfasis en el estudio de sistemas experimentales modelo en los cuales las condiciones de la reacción son mucho más simples y mejor caracterizadas que durante la operación de una celda de combustible. Estos estudios se enfocan principalmente en el análisis del desempeño de orientaciones cristalinas específicas y en metales promotores sobre superficies policristalinas bien definidas. Del mismo modo, se han establecido relaciones y dependencias de la tolerancia al CO con respecto a las principales variables de proceso como la temperatura, la presión y el contenido de CO en el gas alimentado, entre otras.El otro tipo de estudios (una cantidad mucho más reducida que los estudios experimentales) trata de estudiar teóricamente el fenómeno de envenenamiento y el mecanismo de ataque del CO sobre el platino. Producto de tales investigaciones son algunos modelos y simulaciones que intentan describir la situación y que proponen posibles causas estructurales y electrónicas del mismo (Christoffersen et al., 2001; Petrova y Yacovkin, 2002 y 2005).
Este artículo es producto de una revisión bibliográfica, basada en información reportada durante los últimos años y su objetivo es situar al lector en el estado del arte de la investigación sobre el manejo del CO presente en el compartimiento anódico de celdas de combustible.
Es importante resaltar que debido a la gran cantidad de trabajos publicados sobre este tema, es imposible realizar una revisión bibliográfica exhaustiva sin dejar de lado algunos trabajos que se puedan considerar relevantes. Sin embargo, se espera que esta revisión oriente suficientemente al lector sobre el panorama general y el estado de avance de este tipo de estudios. A partir de este trabajo también se pretende mostrar que aun quedan muchos aspectos no aclarados de cuyo conocimiento se podría sacar gran provecho para llegar a nuevos desarrollos en la producción de mejores catalizadores y al aumento de la eficiencia de las celdas.
2. RESULTADOS EXPERIMENTALES
El monóxido de carbono es quizás la molécula cuyas propiedades de adsorción se han investigado con mayor detalle en los sistemas modernos de alto vacío (LEED, AES, XPS). En la actualidad está prácticamente aceptado (Markovic y Ross, 2002) que la adsorción química de CO en metales de transición ocurre en forma molecular a través de la transferencia electrónica del orbital 5σ del CO hacia el metal y de una donación inversa de electrones en el orbital d del metal hacia el anti-enlace 2π* del CO, la cual debilita el enlace C=O. La magnitud de esta donación se incrementa a medida que se consideran metales más a la izquierda de la tabla periódica (Ni, Pd, Pt). Este modelo predice correctamente la adsorción de CO en metales dependiendo de su configuración electrónica.
Estudios de la adsorción de CO sobre diferentes catalizadores establecen que la velocidad de adsorción depende de la naturaleza del catalizador y puede darse por cualquiera de dos rutas (Lee, et al. 1999):
La primera de dichas rutas es una reacción de reemplazo o desplazamiento, en la cual el CO desplaza al hidrógeno atómico adsorbido y ocupa su lugar en la superficie (Adams et al., 2005):
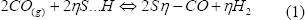
Siendo S los sitios libres de la superficie.
La segunda ruta de adsorción de CO consiste en un ataque a sitios libres:
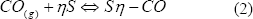
De acuerdo con ese estudio, la reacción (1) es propia de catalizadores PtSn/C, en tanto que la reacción (2) caracteriza la adsorción sobre electrodos Pt/C y PtRu/C.
Sea cual sea la ruta de ataque del CO, el problema que dicho ataque lleva consigo es una desactivación del catalizador. Adicionalmente, el elevado potencial de oxidación del CO sobre Pt dificulta la remoción del contaminante y la consecuente liberación de sitios activos para la posterior oxidación del combustible.
Se han publicado una gran cantidad de trabajos experimentales de la adsorción de CO en superficies modelo de platino: superficies con una estructura cristalina específica en las cuales el CO es previamente disuelto en soluciones acuosas de ácido sulfúrico y posteriormente se permite la adsorción. Por lo general en estas investigaciones se emplean superficies metálicas extendidas y no se consideran las interacciones con el soporte.
Como resumen de esas investigaciones se puede decir que en superficies de platino con índices de Miller bajos, el CO se adsorbe con el carbono enlazado perpendicularmente a la superficie, ocupando principalmente sitios tope y en menor proporción sitios puente (bridge).
En superficies Pt (111), la entalpía de adsorción de CO varía principalmente con el grado de cubrimiento de la superficie debido a interacciones laterales repulsivas entre las moléculas de CO. Adicionalmente, la entalpía de adsorción se ve afectada en una mucho menor medida por la presencia de sitios diferentes de adsorción (escalones, defectos, etc.). La entalpía de adsorción a bajos cubrimientos es aproximadamente 140 kJ/mol. Cuando se tienen altos cubrimientos de CO (cerca al cubrimiento de saturación de 0.68 CO/Pt), este valor disminuye hasta 40 kJ/mol, presumiblemente debido a las fuertes interacciones repulsivas de las moléculas de CO a altos cubrimientos. Una entalpía de adsorción de sólo 40 kJ/mol está en la transición entre quimi-adsorción y fisi-adsorción es por esto que en algunas investigaciones se ha utilizado el término débilmente adsorbido para referirse al estado del CO en los cubrimientos más altos.
Por otra parte, se han postulado dos mecanismos para la oxidación de CO en Pt(h k l): en el primero de ellos se postula que el CO reacciona con moléculas de OH adsorbido mediante una reacción Langmuir-Hinshelwood (L-H). (Ross, P.N, 1998; Markovic et al., 1999; Markovic y Ross, 2002).
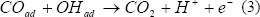
Las especies COad y OHad pueden competir o no por los mismos sitios de la superficie del platino: para CO débilmente adsorbido, se presenta una adsorción no competitiva. Cuando el CO se encuentra fuertemente adsorbido, se presenta una adsorción competitiva por los sitios de adsorción, lo que se manifiesta en un orden de reacción negativo con respecto al CO.
Aunque el mecanismo descrito anteriormente es el más ampliamente aceptado, también se han propuesto modelos diferentes para la oxidación de CO (Bergelin, et al., 1998) los cuales sugieren que para CO débilmente adsorbido, la reacción no procede mediante el mecanismo L-H sino a través de un mecanismo Eley-Riedeal (E-R). Es decir, la reacción se presenta entre el CO y moléculas de agua activadas presentes en la doble capa eléctrica. El debate sobre cual de estos mecanismos es el que opera continúa abierto.
Por otra parte, la relación entre la morfología de los electrodos de platino puro y la tolerancia al CO es un asunto bastante complejo de analizar debido a que desde hace algún tiempo se observaron oscilaciones en la reacción de oxidación de CO en platino (Beusch et al., 1972).
En la primera investigación experimental detallada que se conoce al respecto, se mostró que las oscilaciones en la oxidación de CO en platino están vinculadas con la reestructuración de la superficie, la cual es inducida por el mismo adsorbato (Ertl et al., 1982).
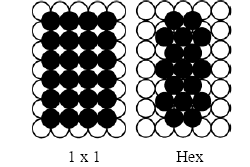
Figura 1. Esquema de la superficie de platino (100). Los círculos en blanco y negro representan la primera y segunda capa de platino, respectivamente.
Figure 1. Sketch of platinum (100) surface. White and Black circles represent the first and second platinum layers, respectively.
Los estudios para la adsorción de CO en Pt (100) muestran que existe una reconstrucción del arreglo cuasi-hexagonal (hex), llevando a un arreglo (1x1) en presencia de CO. Es decir, en una superficie limpia de platino, el arreglo (1x1) de los átomos metálicos (Fig. 1) es meta-estable comparado con el arreglo cuasi-hexagonal (hex). La diferencia energética entre átomos de platino en estos dos arreglos es de aproximadamente 5 kcal/mol. Sin embargo, la adsorción de CO puede estabilizar el arreglo (1 x 1).
También se ha establecido que la exposición al CO de platino (110) con estructura (1x2) lleva a una reconstrucción de la superficie hacia una estructura (1x1) (Fig. 2). Se ha sugerido (Sharma, et al., 1998) que la fuerza impulsora para las reconstrucciones del Pt (100) y Pt(110) es el incremento en el calor de adsorción de CO en estructuras (1x1) el cual excede la diferencia de energía de las superficies limpias. Por ejemplo, a bajos cubrimientos el calor de adsorción de CO en Pt(100)-hex es 106 kJ/mol mientras que en la fase (1x1) es de 156 kJ/mol.
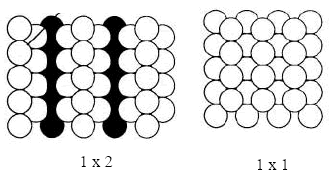
Figura 2. Esquema de la superficie de platino (110). Los círculos en negro representan la primera capa de platino.
Figure 2. Sketch of platinum (110) surface. Black circles represent the first platinum layer.
Oscilaciones en la reacción de oxidación de CO se han hallado en todos los tipos de catalizadores de platino incluyendo partículas soportadas de escala nanométrica, estructuras policristalinas y cristales con orientaciones específicas, en condiciones de operación que van desde alto vació hasta condiciones atmosféricas. En particular, este fenómeno se ha estudiado experimentalmente con detalle en superficies de platino (100) y (110) (Imbihl y Ertl, 1995). Aunque se creía que las superficies de platino con orientación (111) no presentan comportamiento oscilatorio, se ha mostrado que el fenómeno si puede ocurrir (Colen et al., 1998).
Se ha encontrado evidencia experimental (Zhdanov, 2002) de la formación de islas del arreglo (1x1) durante la adsorción de CO en Pt inclusive con bajos niveles de cubrimiento. La cinética de desorción térmica de CO en platino es también un indicativo de que hay una separación de fase en las capas adsorbidas. Estos hallazgos y muchas otras evidencias experimentales indican que en este sistema la reestructuración de la superficie de platino, inducida por el CO adsorbido, puede ser descrita en términos de la teoría de transición de fases de primer orden.
Existen también estudios de la adsorción y oxidación de CO sobre superficies definidas de platino con el suministro de CO en fase gaseosa y en presencia de oxígeno (estos estudios están principalmente enfocados para aplicaciones de control de la polución). En presencia de CO gaseoso, también se presenta una reconstrucción de las superficies Pt(100) y Pt(110) (Imbihl y Ertl, 1995). Esto produce oscilaciones en la reacción de oxidación de CO debido a que la velocidad de adsorción de oxígeno en el arreglo (1 X 1) es mucho más alta que en el arreglo hex. Es decir, la relación de coeficientes de adsorción para CO y O2 cambia dramáticamente con la transformación del arreglo de los átomos de platino.
El comportamiento oscilatorio de las reacciones de oxidación de CO sobre platino también se ha podido corroborar con la detección de un comportamiento oscilatorio en la densidad de estados locales alrededor del nivel de Fermi de los electrones del platino Ds(Ef). Esto ha dado lugar a que en algunas simulaciones moleculares de la adsorción de CO en partículas de platino se utilicen potenciales efectivos de interacción lateral indirecta de la forma:
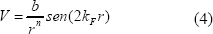
En la cual V es el potencial de interacción lateral, kF es el vector de onda de Fermi y r es la distancia entre la molécula de CO y el sitio de platino.
Con el uso de este tipo de potenciales de interacción se ha logrado una descripción molecular más realista de los procesos de adsorción y oxidación de CO en catalizadores de platino (Petrova y Yacovkin, 2002 y 2005).
Para el caso de la adsorción de CO en partículas de platino soportado (clusters), se han publicado una gran cantidad de trabajos experimentales, en los cuales se reporta el efecto de la morfología sobre la actividad catalítica, mostrándose el platino rugoso con mucha mayor actividad que el platino liso (Mukerjee, 1990). Este crecimiento de la actividad del platino rugoso se ha asociado (en soluciones de ácido sulfúrico) con una disminución del HSO4- fuertemente adsorbido (Armand y Clavilier, 1989, Tripkovic et al., 1998) y con la presencia de escalones y bordes en las partículas los cuales se han vinculado con las primeras fases de adsorción del CO y el agua. La morfología del platino soportado en carbón también cambia con el uso, y algunos investigadores han reportado una pérdida del área superficial con el tiempo (Yu et al., 2005).
Para los aglomerados de platino soportado en grafito, algunos estudios de NMR han permitido detectar un pico agudo para la densidad de estados locales alrededor del nivel de Fermi de los electrones Ds(Ef), lo cual pone en evidencia la fuerte interacción entre los átomos de platino y el soporte, indicando que la adsorción de CO no modifica la densidad de estados del platino y se mantiene la magnitud de las interacciones platino-soporte (Tong et al., 1999). Aunque se ha detectado un incremento en la densidad de estados para las partículas más pequeñas que 5 nm, se sigue considerando que hay una fuerte interacción Pt-C inclusive en esos casos (Tong et al., 2002).
Por otro lado, también se ha demostrado que el método de preparación del catalizador soportado tiene gran influencia sobre su capacidad de adsorción y oxidación de CO (Götz y Wendt, 1998; Xue et al., 2006; Alcaide et al., 2006), siendo preferible, por ejemplo, el método de preparación coloidal propuesto por Bönneman et al. (1991) que el de impregnación. Pero no se presenta ninguna hipótesis que explique esta mejora.
Por esa misma época se presentó un estudio más amplio (Massong et al., (1998), en el cual se muestra el efecto de la depositación a subpotencial de superficies de platino, encontrando que con este modo de preparación, las superficies Pt (332) presentan mayor tolerancia al CO que las superficies Pt (111).
Posteriormente, Brankovic et al. (2001), encontraron que el método de preparación del electrodo puede manipularse de forma tal, que se puedan disminuir las cargas de catalizador. A partir del método propuesto por estos investigadores, se pueden obtener electrodos con baja carga de Pt y alta tolerancia al CO.
3. ESTRATEGIAS DE SOLUCION AL PROBLEMA DEL ENVENENAMIENTO
Del análisis de los trabajos experimentales publicados se pueden dilucidar cinco tendencias principales para manejar el problema de la presencia de CO en el ánodo de las celdas de combustible: a) disminución del potencial de inicio de la reacción de oxidación, b) reducción de la cantidad de CO adsorbido, c) utilización de pequeñas cantidades de oxígeno en la corriente de alimentación al ánodo, d) aumento de la temperatura de operación y e) limpieza del CO a la entrada. A continuación se revisará brevemente cada una de esas tendencias.
3.1 Disminución del potencial de inicio de la reacción
Se ha encontrado que varios metales tienen actividad co-catalítica con el platino. Es decir, que sobre su superficie, el potencial de inicio para la oxidación de CO es lo suficientemente bajo como para que la remoción sea factible, manteniendo de este modo el rendimiento de la celda.
Así, en los últimos años, la gran mayoría de las investigaciones se han orientado hacia la obtención y preparación de electrodos basados en estos materiales (o aleaciones que los contengan) y hacia la caracterización cinética de los electrodos construidos, ya sea individualmente o como componentes de una celda completa.
De entre los metales tolerantes al CO, el rutenio fue tal vez el primero que al combinarse con Pt presentó alta tolerancia al CO. Aun hoy es una de las aleaciones más usadas debido a su excelente desempeño, comparado con muchos otros metales y aleaciones.
Qi et al., (2002) reportaron el efecto de la carga de catalizadores PtRu sobre el desempeño de 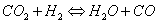 celdas de combustible poliméricas con una alimentación al ánodo compuesta de 10 ppm CO en una corriente de 70% H2 y 30 % CO2. Se encontró que cargas altas de catalizador aumentan la tolerancia al CO.
celdas de combustible poliméricas con una alimentación al ánodo compuesta de 10 ppm CO en una corriente de 70% H2 y 30 % CO2. Se encontró que cargas altas de catalizador aumentan la tolerancia al CO.
Si bien los electrodos de Pt/Ru cuentan con menores potenciales de inicio para la oxidación de CO (~0.45 V) que el Pt puro, varios autores (Tacconi et al., 1994; Grgur et al., 1999; Murkejee et al. 1999; Orozco y Gutiérrez, 2000) han señalado la posibilidad de obtener menores potenciales de inicio y mejores desempeños usando otros metales como el Re, Mo, Sn, Ir y Os, entre otros.
De estas investigaciones vale la pena resaltar el trabajo de Grgur et al. (1998 y 1999). Ese trabajo consistió en el estudio de la electro oxidación de H2, CO y mezclas H2/CO sobre un catalizador Pt75/Re25, para el cual encontraron excelente actividad catalítica para oxidación de H2 y de CO puros. Sin embargo, para mezclas de los dos gases, que es el caso de interés, el potencial de inicio de la reacción resultó ser mayor que el de electrodos Pt/Ru y Pt3Sn. Adicionalmente a esto, las densidades de corriente obtenidas a bajos potenciales fueron muy pequeñas (sólo 3(A/cm2).
Posteriormente, se presentaron otros estudios en los cuales se probó al molibdeno como acompañante del platino (Murkejee et al. 1999). Los resultados hallados establecen que en los catalizadores PtMo la oxidación de CO comienza a potenciales muy bajos (~0.1 V) y es debida a especies intermedias oxidadas del Mo. A partir de estos estudios, se encontró que el Mo es casi tan buen catalizador para la oxidación de H2 puro como el Pt, con la ventaja adicional de que la actividad de electrodos PtMo para la oxidación de CO es muy superior a la de electrodos de Pt. Además, dicha actividad alcanza a ser de dos a tres veces mayor a la de electrodos PtRu a bajos potenciales y condiciones típicas de operación de una celda de combustible ( 85°C y 100 ppm de CO). Específicamente, para aleaciones PtMo en relación atómica 4:1 y concentraciones de CO de 100 ppm, se encontró entre 20 y 25% de incremento en el potencial pico de oxidación de CO, respecto al presentado con mezclas PtRu.
Papageorgopoulos et al.( 2002 a) encontraron que la adición de 10% de Mo en mezclas Pt-Ru produce un catalizador con mayor actividad (que la mezcla original Pt-Ru) cuando se tiene presencia de CO. Posteriormente Urian et al., (2003) investigaron la tolerancia al CO y CO2 de cuatro tipos de PtMo/C (relaciones atómicas Pt:Mo de 1:1, 3:1, 4:1 y 5:1) y las compararon con las respuestas obtenidas con Pt y PtRu (1:1). En ese estudio se reporta que los catalizadores PtMo presentan al menos tres veces mayor tolerancia al CO que los catalizadores PtRu y que la relación atómica Pt:Mo no tiene prácticamente ninguna influencia en la polarización de CO cuando este se encuentra en una concentración de 100 ppm.
Aunque esos resultados muestran que el PtMo es un catalizador anódico promisorio, tiene una desventaja que ha detenido su aplicación practica: Ball et al. (2002) mostraron que aunque la mezcla PtMo tiene alguna ventaja sobre la mezcla PtRu para la tolerancia al CO, en presencia del CO2 (el otro componente principal de la corriente de reformado), el desempeño de PtMo es significativamente inferior al mostrado por PtRu. La razón de este pobre desempeño del catalizador PtMo en presencia de CO2 se atribuyó al aumento de la reacción inversa de desplazamiento de agua, catalizada por el PtMo (la cual consume hidrógeno):
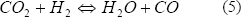
Por otra parte, las mezclas Pt-Sn también se han estudiado con un interés considerable (Arico et al., 1994; Morimoto y Yeager, 1998; Kelaidopoulou et al., 1999). Una caracterización extensiva mediante microscopio electrónico de transmisión (TEM), análisis de energía dispersiva de rayos X (EDX), espectroscopia de rayos X (XPS) y voltametría cíclica (CV) (Crabb et al., 2000) muestra que se pueden obtener mezclas PtSn con tamaño de partícula de 3 a 4 nm con un método de preparación órgano-metálico. Una vez esta mezcla se expone al aire, se producen óxidos de platino y de estaño. El potencial de inicio de la reacción de oxidación de CO se desplazó hacia potenciales más bajos (alrededor de 400 mV comparado con Pt).
También se ha buscado mejorar los catalizadores a partir de la combinación de tres o más metales: Pt-Ru-Os, Pt-Ru-Rh, Pt-Ru-Sn, Pt-Ru-Sn-W etc (Yang et al., 2004; Vigier et al., 2004). Con esto se pretende capitalizar las propiedades de cada uno de los metales, optimizando el comportamiento de la mezcla.
Götz y Wendt (1998) probaron formulaciones binarias y ternarias de catalizadores que incluyen metales como el W, Sn y Mo para celdas de combustible poliméricas (PEMFCs) operadas con metanol y gas reformado. Dichos catalizadores se compararon con uno de PtRu, el cual sirvió como patrón de referencia. Los investigadores mostraron que con las preparaciones trimetálicas basadas en Pt/Ru, se presentaron dos resultados: para bajas corrientes el mejor catalizador fue el de Pt/Ru/W, mientras que para altas corrientes, presentó mejor desempeño el de Pt/Ru/Sn.
Muchas otras formulaciones ternarias se han publicado (Chunzi et al. 2003; He et al., 2003; Venkataraman et al, 2003). En estos trabajos se estudiaron las mezclas Pt/Ru, Pt/Ru/Au, Pt/Ru/Os, Pt/Ru/SnOx y Pt/Ru/WOx, A partir de las pruebas realizadas se encontró que el catalizador de Pt/Ru/WOx tiene aproximadamente el doble de actividad que el de Pt/Ru para electro oxidación de mezclas H2/1%CO, presentando menor potencial de inicio para la reacción que este último. También se encontró que el electrodo de Pt/Ru/SnOx tuvo el menor potencial de inicio para oxidación de CO, pero la cinética de reacción fue baja. El electrodo de Pt/Ru/Au tuvo el peor desempeño.
Otro grupo (Venkataraman et al 2003, 2004a, 2004b) estudió la influencia de aleaciones de Pt/Ru/Ag, Pt/Ru/Au, Pt/Ru/Rh y Pt/Ru/W2C sobre la electro-oxidación de mezclas H2/CO. Los resultados obtenidos indicaron que la presencia de estos componentes reduce la cantidad de Ru aleado con Pt en el catalizador e incrementa el potencial de oxidación de CO. Este mismo grupo reporta el uso de complejos aminos de metales de transición como co-catalizadores de mezclas H2/CO. Se cree que estos complejos actúan como mediadores redox en la generación de especies que oxidan al CO. Los autores también observaron que la síntesis de catalizadores de platino utilizando Na2S2O3 puede llevar a la generación un nuevo tipo de catalizadores de platino conteniendo sulfuro los cuales muestran inhibición a la adsorción de CO y potenciales de oxidación de CO tan bajos como 0.2 V.
Aunque varios de los electrodos ternarios tienen potenciales de inicio para la reacción de oxidación de CO menores que la mezcla PtRu, aquellos tienen problemas asociados a su método de preparación, a su estabilidad en el tiempo y a su bajo desempeño.
Actualmente se sigue considerando a la mezcla Pt-Ru como el electrocatalizador anódico más eficiente para combustibles que contienen 10 ppm de CO o más (Gasteiger et al., 2005; Papageorgopoulos et al., 2002 a), debido a los efectos electrónicos y bifuncionales que reducen el envenenamiento y por lo tanto el desarrollo de catalizadores tolerantes al CO todavía está concentrado en sistemas basados en Pt o en Pt-Ru. Sin embargo, cuando se emplea hidrógeno puro, una celda de combustible con un ánodo de Pt-Ru tiene un más bajo desempeño que una de Pt (Acres et al., 1997). De hecho, en este caso el uso de Pt-Ru resulta en una pérdida sustancial en el voltaje de la celda (Iorito et al., 2003; Adams et al., 2005).
Todo esto pone de manifiesto que se requiere un trabajo investigativo adicional para la producción de catalizadores con bajo potencial de inicio para la reacción de oxidación de CO, alta actividad catalítica, estabilidad y flexibilidad en cuanto a la concentración del CO en la corriente de alimentación.
3.2 Reducción de la cantidad de CO adsorbido
Para algunos metales el mecanismo de tolerancia al CO está basado en impedir o reducir su adsorción sobre el catalizador, evitando de este modo el bloqueo de los sitios activos. El paladio es un claro ejemplo de este tipo de tolerancia al envenenamiento (Pyun et al., 1996; Papageorgopoulos et al., 2002b). Este último grupo estudió los efectos del envenenamiento con CO sobre electro catalizadores Pt/Pd (ricos en Pd) mediante voltametría cíclica y pruebas en PEMFCs.
Los resultados obtenidos de las voltametrías de despojamiento de CO, demuestran que la adición de Pd al catalizador disminuye el cubrimiento de CO en la superficie del mismo. Incrementos en la cantidad de Pd adicionada hacen más pronunciado el efecto. Esta reducción del cubrimiento, es decir, el hecho de que se adsorba menor cantidad de CO sobre la superficie del catalizador, deja fracciones libres de sitios activos donde se lleva a cabo la oxidación de H2. Como consecuencia, el catalizador obtenido es más tolerante al CO, pues no puede ser envenenado fácilmente.
En cuanto a las pruebas en celdas de combustible, el electrodo PtPd4 presentó una mayor tolerancia al CO que el de PtRu, con concentraciones de CO de 100 ppm o mayores en la corriente de combustible. Infortunadamente, las densidades de corriente obtenidas utilizando Pd son muy bajas como para pensar en desarrollos comerciales con este tipo de celdas.
También se han probado las sistemas Pd-Au/C (Schmidt et al., 1998 y 2001), para el cual se propone que el Pd proporciona un medio altamente activo a la oxidación de hidrógeno y el segundo metal (Au) produce superficies que no adsorben CO bajo las condiciones de operación de la celda, resultando que para bajos sobrepotenciales y temperatura ambiente, existen más sitios de adsorción para hidrógeno en PdAu que sobre PtRu. A 60°C la superficie PdAu apareció menos envenenada que la mezcla PtRu. Sin embargo, estos resultados requieren mayor estudio, especialmente en cuanto al tamaño de partícula, la homogeneidad y la cantidad que se requiere de cada metal en la mezcla.
3.3 Utilización de pequeñas cantidades de oxígeno en la corriente de alimentación al ánodo
Algunos autores han propuesto la utilización de pequeñas cantidades de oxígeno en la corriente de alimentación a la celda con miras a ayudar a la oxidación de CO y reducir el efecto del envenenamiento (Schmidt et al., 1997; Gottesfeld y Pafford, 1998; Bellows et al., 1998).
Se ha mostrado que la tolerancia al CO en el ánodo se puede incrementar significativamente cuando se inyectan bajas concentraciones de oxígeno o de aire (Gottesfeld y Pafford, 1998) y se reporta que con una inyección de un 4.5% de O2 en una corriente de H2/100 ppm CO el desempeño de la celda se recupera completamente a 85 °C. Es decir, a estas condiciones la celda se desempeña igual que con hidrógeno puro.
Murthy et al., (2001) estudiaron el efecto de alimentar aire sobre la respuesta de una celda polimérica expuesta a concentraciones relativamente altas de CO (entre 500 y 3000 ppm.). Encontraron que alimentar un 5% de aire puede producir una buena tolerancia para 500 ppm de CO.
Una leve variación de este método es la introducción de H2O2 en el humidificador del ánodo (Schmidt et al., 1997). Según este método, un 5% de H2O2 en el humidificador puede mitigar el envenenamiento de una corriente de hidrógeno con 100 ppm de CO.
Dentro de esta estrategia también se ha probado la modificación de la estructura del ánodo (Rohland y Plzak, 1999) mediante una espuma metal/grafito que se pone a la entrada de la corriente de gas y en la cual se deposita una mezcla de platino y oro, esto mejora la tolerancia al CO hasta 750 ppm con una muy baja inyección de oxígeno.
Aunque la estrategia de inyectar oxígeno al ánodo parece promisoria, no parece ser completamente beneficiosa debido a que en este caso también se presenta la reducción de oxígeno, produciendo H2O2 el cual deteriora rápidamente la membrana y algunos investigadores han planteado inconvenientes de seguridad operando con esta técnica (Hou, et al., 2003; Batista, et al., 2005).
3.4 Aumento de la temperatura de operación
En general puede decirse que los incrementos de temperatura favorecen la tolerancia al CO de los diferentes catalizadores usados en electrodos (ánodos) de celdas de combustible. Este comportamiento obedece a que la cinética de oxidación de CO sobre las superficies de catalizadores, es más rápida a temperaturas más altas y a que la energía del enlace Pt-CO disminuye (Schmidt et al., 1999; Kim et al., 2001).
En general, a temperaturas elevadas, la fortaleza del enlace de CO sobre Pt disminuye notablemente, con lo cual se favorece su desorción. De hecho, por encima de 200°C, esta fuerza de enlace se hace muy similar a la del enlace CO sobre oro a 80°C (Rohland y Plzak, 1999). A dicha temperatura, la tolerancia del oro al envenenamiento por CO es muy alta, por lo cual la comparación anterior permite hacerse una idea del mejoramiento de la tolerancia al envenenamiento con incrementos de temperatura.
Lee et al., (1999) también mostraron el efecto de la temperatura en el desempeño de celdas de combustible alimentadas con H2 y 20 ppm de CO. Mostraron que el efecto del envenenamiento es mucho más severo a bajas temperaturas. Reportan una densidad de corriente de 1.5 A/cm2 a 0.4 V operando a 115°C.
Operar la celda a altas temperaturas puede incrementar la tolerancia al CO. Sin embargo, la mayoría de las membranas de electrolito polimérico disponibles en la actualidad no operan apropiadamente por encima de 100 °C debido a la deshidratación de la membrana, la pérdida de conductividad protónica y la pérdida de estabilidad termo-mecánica del polímero. La evaluación del envenenamiento con CO en celdas de combustible operando hasta 200°C empleando membranas de PBI dopadas con acido fosfórico (Li et al., 2003) muestra que se puede tolerar un 3% de CO en H2 para densidades de corriente hasta de 0.8 A/cm2.
Existe una gran cantidad de esfuerzo investigativo en desarrollar membranas de intercambio protónico nuevas o modificadas para la operación a temperaturas mayores a 120 °C. (Molhotra y Datta, 1997; Yang et al., 2001; Adjemian et al., 2002; Xu et al., 2006), pero la revisión detallada de esas investigaciones está más allá del alcance de este trabajo.
3.5 Limpieza del CO a la entrada
Otra manera de evitar el envenenamiento del catalizador consiste en limpiar el combustible antes de que llegue al ánodo.
Mediante esta estrategia se adicionan dispositivos externos a la celda con catalizadores Rh/MgO, Ru/γ-Al2O3 y Pt/ γ-Al2O3 (Han et al., 2004; Wakita et al., 2006); Rh-K/ γ-Al2O3 (Chen et al., 2004); Au/CeOx (Panzera et al., 2004) y Ce-Pt/ γ-Al2O3 (Son, 2006) entre otros. De entre estos catalizadores, el Rh-K/ γ-Al2O3 parece ser el más promisorio.
Infortunadamente, como es bien sabido, la adición de dispositivos externos a la celda resulta engorrosa y costosa, sobre todo si se quieren alcanzar niveles aceptables de CO para evitar envenenamiento. Inclusive con la adición de dispositivos externos de limpieza es difícil mantener bajos niveles de CO durante el periodo de encendido o en regimenes de operación cambiantes (Batista et al., 2004; Heinzel et al., 2002).
Es razonable entonces pensar en implementar un dispositivo al interior de la celda que realice una limpieza del H2, previamente a su llegada al catalizador de Pt. Por otra parte, se simplifica el montaje al ubicar el dispositivo de limpieza al interior de la celda.
Rohland y Plzak (1999), proponen el uso de una hoja catalizadora basada en oro, la cual se ubicaría a la entrada del gas alimentado al ánodo. Esta hoja logra una oxidación selectiva del CO, permitiendo el paso de hidrógeno hacia el resto del catalizador.
Empleando una idea semejante, Huang et al., (2002a y 2002 b) pusieron una capa de rutenio (al cual llamaron filtro) en la parte más externa del ánodo para recibir la corriente con CO. Se postula que el filtro de rutenio promueve la reacción de oxidación de CO, ya que la cinética de reacción de H2 en rutenio es muy baja.
Yu et al., (2002) proponen un electrodo compuesto en el cual la capa adyacente a la entrada de los gases se compone de una mezcla PtRu (la cual se emplea para catalizar la oxidación de CO) y de una capa más interna compuesta de Pt (para catalizar la oxidación de hidrógeno). En la configuración propuesta la capa más externa contiene la mayor cantidad del catalizador, mientras que internamente se utiliza una capa delgada con mucho menor cantidad de catalizador. Esto representa un mejoramiento aparente sobre la estructura convencional.
Más recientemente, (Santiago et al., 2004) proponen el montaje de un catalizador Mo/C en la capa de difusión del ánodo y empleando Pt/C, PtRu/C or PtMo/C en la capa catalítica. Esta estructura presentó un aumento de la actividad catalítica a la oxidación de hidrógeno en presencia de CO, la cual fue atribuida a una más baja concentración de CO que llega al catalizador debido a la reacción de desplazamiento de agua, la cual es catalizada por el Mo/C.
En general, debido a los diferentes coeficientes de difusión del CO y el H2, es posible diseñar electrodos con estructuras de múltiples capas que tengan en cuenta los diferentes componentes del combustible. Cada una de estas capas puede tener diferentes tipos de catalizador y de estructura porosa y aunque este tipo de estrategia no ha recibido la mayor atención, puede llegar a ser una alternativa muy atractiva para la disminución del envenenamiento en celdas de combustible.
4. CONCLUSIONES
Se pueden identificar cinco tendencias principales para reducir el envenenamiento del catalizador anódico en celdas de combustible cuando se tiene presencia de CO: a) disminución del potencial de inicio de la reacción de oxidación, b) reducción de la cantidad de CO adsorbido, c) utilización de pequeñas cantidades de oxígeno en la corriente de alimentación al ánodo, d) aumento de la temperatura de operación y e) limpieza del CO a la entrada.
De estas estrategias, la primera es la que ha recibido considerablemente mayor atención y aunque se han desarrollado catalizadores bastante promisorios (especialmente PtMo y PdAu), todavía se sigue considerando a la mezcla Pt-Ru como el electrocatalizador anódico más eficiente para combustibles que contienen 10 ppm de CO o más. De todas maneras se requiere un trabajo investigativo adicional para la producción de catalizadores con bajo potencial de inicio para la reacción de oxidación de CO, alta actividad catalítica, estabilidad y flexibilidad en cuanto a la concentración del CO en la corriente de alimentación.
La estrategia de inyectar oxígeno al ánodo parece promisoria pero requiere el desarrollo de nuevas membranas más resistentes y de la implementación de condiciones más seguras de operación de la celda.
El diseño estructural de ánodos especiales que contengan capas con catalizadores específicos para cada tipo de combustible puede ser una estrategia muy atractiva para la disminución del envenenamiento en celdas de combustible. Dentro de esta estrategia todavía hay mucho campo de estudio en cuanto a la optimización de la estructura para hacer más efectiva la tolerancia al CO.
Es claro que la gran mayoría de la investigación en torno a los procesos de adsorción y oxidación de CO en platino está basada en una aproximación principalmente experimental. Por esta vía se han invertido una gran cantidad de recursos con la esperanza de obtener en un plazo relativamente corto catalizadores con tolerancia suficientemente aceptable al CO como para pensar en la comercialización de celdas de combustible.
Aunque algunos de los catalizadores desarrollados por la vía experimental presentan propiedades atractivas, esta forma de abordar el problema se debe combinar con una estrategia diferente de búsqueda de nuevos catalizadores, la cual debe consistir en trabajos teóricos para descartar las combinaciones de posible menor desempeño para así reducir el tiempo y los esfuerzos experimentales.
El modelamiento teórico es probablemente en estos momentos el camino más conveniente para entender los detalles de los fenómenos moleculares que ocurren en la superficie del platino y puede generar un mayor entendimiento de la manera en que la morfología y la distribución de tamaño de las partículas de platino influyen en la adsorción y oxidación de CO.
REFERENCIAS
[1] ACRES, G, J., J.C. FROST, G.A. HARDS, R.J. POTTER, T.R. RALPH, D. THOMPSETT, G.T. BURSTEIN, G.J. HUTCHINGS, Catal. Today, 38 393–400, 1997. [ Links ]
[2] ADAMS, W. A., J. BLAIR, K. BULLOCK, C. GARDNER, J. Power Sources, 145, 55–61, 2005. [ Links ]
[3] ADJEMIAN, K., S. J. LEE, J. BENZIGER, AND A.B. BOCARSLY, J. Electrochem. Soc., 149, A256, 2002. [ Links ]
[4] ALCAIDE, F., O. MIGUEL, H-J. GRANDE, Catal. Today, 116, 408–414, 2006. [ Links ]
[5] ARICO, A. S., V. ANTONUCCI, N. GIORDANO, A. K. SULA, M. R. RAVIKAMAR, A. ROY, S. R. BARMAN Y D. D. SARMA, J. Power Sources, 50, 295-309, 1994. [ Links ]
[6] ARMAND, D., Y J. CLAVILIER, J. Electroanal. Chem., 263, 109, 1989. [ Links ]
[7] BALL, S., A. HODGKINSON, G. HOOGERS, S. MANIGUET, D. THOMPSETT, Y B. WONG, Electrochem. Solid-State Lett., 5, A31, 2002. [ Links ]
[8] BATISTA, M.S., E.I. SANTIAGO, E.M. ASSAF, E.A. TICIANELLI, J. Power Sources, 145, 50–54, 2005. [ Links ]
[9] BATISTA, M. S., E.I. SANTIAGO, E.M. ASSAF, E.A. TICIANELLI, J. Power Sources, 134, 27–32, 2004. [ Links ]
[10] BELLOWS, R., E. MARUCCHI-SOOS, R.P. REYNOLDS, Electrochem. Solid-State Lett., 1, 69–70, 1998. [ Links ]
[11] BERGELIN, M., J. M. FELIU Y M. Wasberg. Electrochim. Acta, 44, 1069, 1998. [ Links ]
[12] BEUSCH, H., P. FIEGUTH, E. WICKLE, Chem. Eng. Technol., 44, 445, 1972. [ Links ]
[13] BÖNNEMAN et al. Angew. Chem. Int. Ed., 30, 1312, 1991. [ Links ]
[14] BRAKO, R. Y SOKCEVIC H. Vacuum, 61, 89-93, 2001. [ Links ]
[15] BRANKOVIC, S.R., J.X. WANG, R.R. ADZIC. Electrochem. Solid-State Lett., 4, 12, A217, 2001. [ Links ]
[16] CHEN, G., QUAN YUAN, HENGQIANG LI, SHULIAN LI, Chem. Eng. J., 101, 101–106, 2004. [ Links ]
[17] CHRISTENSEN, P. A., J. JIN, W. LIN Y A. HAMNETT. J. Phys. Chem. B., 108, 3391-3394, 2004. [ Links ]
[18] CHRISTOFFERSEN, E., P. LIU, A. RUBAN, H. L. SKRIVER, AND J. K. NØRSKOV. J. OF CATAL., 199, 123–131, 2001. [ Links ]
[19] CHUNZHI, HE, H.R. KUNZ, J.M. FENTON, J. Electrochem. Soc., 150, A1017, 2003. [ Links ]
[20] COLEN, R., J. CHRISTOPH, F. PENA, H. Rotermund. Surf. Sci., 408, 310, 1998. [ Links ]
[21] COUTO, A., M. C. PÉREZ, A. RINCÓN Y C. GUTIÉRREZ, J. PHYS. Chem., 100, 19538-19544, 1996. [ Links ]
[22] CRABB, E. M., R. MARSHALL, Y D. THOMPSETT, J. Electrochem. Soc., 147, 4440, 2000. [ Links ]
[23] ERTL, G., P. R. NORTON, J. RUSTIG. Phys. Rev. Lett. 49, I77, 1982. [ Links ]
[24] GASTEIGER, H. A., S.S. KOCHA, B. SOMPALLI, F.T. Wagner, Appl. Catal. B: Environ. 56, 9–35, 2005. [ Links ]
[25] GASTEIGER, H. A., J.E. PANELS Y S.G. YAN. J. Power Sources, 127, 162–171, 2004. [ Links ]
[26] GASTEIGER, H. A, N. MARKOVIC Y P. N. ROSS, J. Phys. Chem., 99, 8290-8301, 1995a. [ Links ]
[27] GASTEIGER, H. A, N. MARKOVIC Y P. N. ROSS, J. Phys. Chem., 99, 8945-8949, 1995b. [ Links ]
[28] GASTEIGER, H.A, N. MARKOVIC Y P. N. ROSS, J. Phys. Chem., 99, 16757-16767, 1995c. [ Links ]
[29] GOTTESFELD, S, Y J. PAFFORD, J. Electrochem. Soc., 135, 2651-2652, 1998. [ Links ]
[30] GÖTZ, M., H. Wendt. Electrochimica Acta., 43, 3637, 1998. [ Links ]
[31] GRGUR, B.N., N.M. MARKOVIC, P.N. ROSS. Electrochim. Acta., 43, 3631, 1998. [ Links ]
[32] GRGUR, B.N., N.M. MARKOVIC, P.N. ROSS. J. Electrochem. Soc., 146, 1613-1619, 1999. [ Links ]
[33] HAN, Y., M.J. KAHLICH., M. KINNE, R.J. BEHM APPL. CATAL. B: Environ., 50, 209–218, 2004. [ Links ]
[34] HAUG, A., R. E. WHITE, J. W. WEIDNER, W. HUANG, S. SHI, N. RANA, S. GRUNOW,T. C. STONER, AND A. E. KALOYEROS, J. Electrochem. Soc., 149, A868, 2002 a. [ Links ]
[35] HAUG, A., R. E. WHITE, J. W. WEIDNER, W. HUANG, J. Electrochem. Soc., 149, A862, 2002b. [ Links ]
[36] HE, C., H. R. KUNZ, Y J. M. FENTON, J. Electrochem. Soc., 150, A1017, 2003. [ Links ]
[37] HEINZEL, A., B. VOGEL, P. HUBNER, J. Power Sources 105, 202– 207, 2002. [ Links ]
[38] HOU, Z, B. YI., H. YU, Z. LIN, H. ZHANG, J. Power Sources, 123, 116–125, 2003. [ Links ]
[39] IMBIHL, R., G. Ertl. Chem. Rev., 95, 697, 1995. [ Links ]
[40] IORIO, T., K. YASUDA, Z. SIROMA, N. FUJIWARA, Y. MIYAZAKI, J. Electrochem. Soc., 150, 1225–1230, 2003. [ Links ]
[41] JUSYS, Z., H. MASSONG, Y H. BALTRUSCHAT, J. Electrochem. Soc., 146, 1093-1098, 1999. [ Links ]
[42] KELEIDOPOULOU, A., E. ABELIDOU Y G. KOKKINIDIS, J. APPL. electrochem., 29, 1255-1261, 1999. [ Links ]
[43] KIM, J. AND PARK, K. KOBAYASHI, M. NAGAI. J. Power Sources. 103, 127, 2001. [ Links ]
[44] LEE, S.J., S. MURKEJEE, E.A. TICIANELLI, J.MC. Breen. Electrochim, Acta., 44, 3283, 1999. [ Links ]
[45] LI, Q., R. HE, J. GAO, J. O. JENSEN, AND N. J. BJERRUM, J. Electrochem. Soc., 150, A1608, 2003. [ Links ]
[46] MARKOVIC, N., B. N. GRGUR, C. A. LUCAS Y P. N ROSs. J. Phys. Chem. B., 113, 487, 1999 [ Links ]
[47] MARKOVIC, N.M. Y P. N. ROSS. SURF. Sci. Reports., 45, 117-229, 2002. [ Links ]
[48] MASSONG, H., S. TILLMANN, T. LANGKAU, E.A. ABD EL MEGUID, H. BALTRUSCHAT. Electrochim. Acta. 44, 1379, 1998. [ Links ]
[49] MOLHOTRA, S. AND R. DATTA, J. Electrochem. Soc., 144, L23, 1997. [ Links ]
[50] MORIMOTO, Y. Y E. B. YEAGER, J. Electroanal. Chem. Vol. 444, pp. 95-100, 1998. [ Links ]
[51] MUKERJEE, S. J. APPL. Electrochem, 20, 537-548, 1990. [ Links ]
[52] MURKEJEE, S., S. LEE, E. TICIANELLI, J. BREEN, B. GRGUR, N. MARKOVIC, P.N. ROSS, J. GIALLOMBARDO, E. DE CASTRO. Electrochem. Solid-State Lett. 2, 12-15, 1999. [ Links ]
[53] MURTHY, M., M. ESAYIAN, A. HOBSON, S. MACKENZIE, W. LEE, AND J. W. VAN ZEE, J. Electrochem. Soc., 148, A1141, 2001. [ Links ]
[54] OETJEN, H., V. M. SCHMIDT, U. STIMMING, Y F. TRILA, J. Electrochem. Soc., 143, 3838, 1996. [ Links ]
[55] OROZCO, G. Y C. GUTIÉRREZ, J. Electroanal. Chem., 484, 64-72, 2000. [ Links ]
[56] PANZERA, G., V. MODAFFERI, S. CANDAMANO, A. DONATO, F. FRUSTERI, P.L. ANTONUCCI,. J. Power Sources, 135, 177–183, 2004. [ Links ]
[57] Papageorgopoulos, D. C., M. Keijzer, F.A. de Bruijn, Electrochim. Acta., 48, 197–204, 2002 [ Links ]
[58] Papageorgopoulos, D. C., F.A. de Bruijn, J. Electrochem. Soc., 149, 140–145, 2002 b. [ Links ]
[59] Papageorgopoulos, D. C., M. Keijzer, J.B.J. Veldhuis, F.A. de Bruijn. J. Electrochem. Soc., 149, A1400, 2002. [ Links ]
[60] PETROVA, N., Y I.N. YAKOVKIN. Surf. Sci., 578, 162–173, 2005. [ Links ]
[61] PETROVA, N., Y I.N. YAKOVKIN. Surf. Sci. 519, 90–100, 2002. [ Links ]
[62] PYUN, S. I., T. H. YANG Y C. S. KIM, APPL. Electrochem., 26, 953-958, 1996. [ Links ]
[63] QI, Z., C. HE, Y A. KAUFMAN, J. Power Sources, 111, 239, 2002. [ Links ]
[64] ROHLAND, B., V. PLZAK. J. Power Sources, 84, 183, 1999. [ Links ]
[65] ROSS, P.N. Electrocatalysis. Lipkowsky y Ross (Eds). Wiley-VCH, New York, 43-74, 1998. [ Links ]
[66] SANTIAGO, E., M. S. BATISTA, E. M. ASSAF, AND E. A. TICIANELLI, J. Electrochem. Soc., 151, A944, 2004. [ Links ]
[67] SHARMA, R. K., W. A. BROWN Y D. A. KING. Surf. Sci., 414, 68, 1998. [ Links ]
[68] SCHMIDT, T. J., Z. JUSYS, H.A. GASTEIGER, R.J. BEHM, U. Endruschat, H. Boennemann, J. Electroanal. Chem., 501, 132–140, 2001. [ Links ]
[69] SCHMIDT, T. J., H.A. GASTEIGER, R.J. BEHM. J. Electrochem. Soc., 146, 1296, 1999. [ Links ]
[70] SCHMIDT, T. J., M. NOESKE, H.A. GASTEIGER, R.J. BEHM, P. BRITZ, H. BONNEMANN, J. Electrochem. Soc. 145, 925–931, 1998. [ Links ]
[71] SCHMIDT, V. M. H.-F. OETJEN, J. DIVISEk, J. Electrochem. Soc., 144, L237–L238, 1997 [ Links ]
[72] SON I. H., J. Power Sources, 159, 1266–1273, 2006. [ Links ]
[73] SPENDELOW, J. S., G.Q. LU, P.J. KENIS Y A. WIECKOWSKI. J. Electroanal. Chem., 568, 215-224, 2004. [ Links ]
[74] ] TACCONI, N. R., R. O. LEZNA, B. BADEN, F. HANH Y C. LAMY, J. Electroanal. Chem., 379, 329-337, 1994. [ Links ]
[75] TONG, Y., C. RICE, N. GODBOUT, A. WIECKOWSKI, Y E. OLDFIELD. J. AM. CHEM. Soc. 121, 2996-3003, 1999. [ Links ]
[76] TONG, Y., P. BABU, A. WIECKOWSKI Y E. OLDFIELD. Chem. Phys. Lett., 361, 183–188, 2002. [ Links ]
[77] TRIPKOVIC, A.V., K. D. POPOVIC, J. D. MOMCILOVIC Y D. M. DRAZIC, Electrochim. Acta, 44, 1135-1145, 1998. [ Links ]
[78] URIAN, R. C., A.F. GULLÁ, S. MUKERJEE, J. Electroanal. Chem. 554–555, 307–324, 2003. [ Links ]
[79] VENKATARAMAN, R., H.R. KUNZ, J.M. FENTON. J. Electrochem. Soc., 150, A278, 2003. [ Links ]
[80] VENKATARAMAN, R., H. R. KUNZ, AND J. M. FENTON, J. Electrochem. Soc., 151, A703 (2004a). [ Links ]
[81] VENKATARAMAN, R., H. R. KUNZ, AND J. M. FENTON, J. Electrochem. Soc., 151, A710 (2004b). [ Links ]
[82] VIGIER, F., C. COUTANCEAU, A. PERRAND, E. M. BELGSIR Y C. LAMY. J. Appl. Electrochem., 34, 439- 446, 2004. [ Links ]
[83] WAKITA H., Y. KANI, K. UKAI, T. TOMIZAWA, T. TAKEGUCHI, W. Ueda. Appl. Catal. A: General 283, 53–61, 2005. [ Links ]
[84] XU, H., Y. SONG, H. R. KUNZ, J. M. FENTON. J. Power Sources, 159, 979–986, 2006. [ Links ]
[85] XUE, X., J. GE, C. LIU, W. XING, T. LU, Electrochem. Commun., 8, 1280–1286, 2006. [ Links ]
[86] YANG, C., P. COSTAMAGNA, S. SRINIVASAN, J. BENZIGER, AND A.B. BOCARSLY, J. Power Sources, 103, 2001. [ Links ]
[87] YANG, L. X., C. BOCK, B. MACDOUGALL Y J. PARK. J. Appl. Electrochem., 34, 427-438, 2004. [ Links ]
[88] YU, H., Z. HOU, B. YI, AND Z. LIN, J. Power Sources, 105, 52-57, 2002. [ Links ]
[89] YU, P., M. PEMBERTON Y P. PLASSE. J. Power Sources, 144, 11, 2005. [ Links ]
[90] ZHDANOV, V. P., Surf. Sci. Reports., 45, 231, 2002. [ Links ]

