










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkDYNA
Print version ISSN 0012-7353On-line version ISSN 2346-2183
Dyna rev.fac.nac.minas vol.77 no.163 Medellín July/Sept. 2010
KINETIC BEHAVIOR IN THE HYDROGENATION OF FURFURAL OVER IR CATALYSTS SUPPORTED ON TIO2
COMPORTAMIENTO CINÉTICO DE LA HIDROGENACIÓN DE FURFURAL SOBRE CATALIZADORES DE IR SOPORTADOS EN TIO2
HUGO ROJAS
Escuela de Ciencias Químicas, Facultad de Ciencias, Grupo de Catálisis (GC-UPTC), Universidad Pedagógica y Tecnológica de Colombia, Tunja, Colombia, hurojas@udec.cl
JOSÉ J. MARTÍNEZ
Escuela de Ciencias Químicas, Facultad de Ciencias, Grupo de Catálisis (GC-UPTC), Universidad Pedagógica y Tecnológica de Colombia, Tunja, Colombia
PATRICIO REYES
Facultad de Ciencias Químicas, Universidad de Concepción, Chile
Received for review November 4th, 2009, accepted December 15th, 2009, final version January, 18th, 2010
ABSTRACT: The kinetics of the liquid-phase hydrogenation of furfuraldehyde to furfuryl alcohol over Ir catalysts supported over TiO2 was studied in the temperature range of 323 to 373 K. The effect of furfural concentration, hydrogen pressure and the solvent effect were also studied. A high selectivity towards furfuryl alcohol was demonstrated. Initial rates describes the order global of the reaction. The experimental data could also be explained using the Langmuir-Hinshelwood model with of a single-site with dissociative adsorption of hydrogen and the surface reaction as the rate-controlling step provided the best fit of the experimental data.
KEYWORDS: Furfural, hydrogenation, SMSI effect, kinetic study.
RESUMEN: La cinética de la hidrogenación en fase líquida de furfural a alcohol furfurilico sobre catalizadores de Ir/TiO2 se estudio en el rango de temperaturas de 323 a 373 K, también se estudio el efecto de la concentración de furfural, presión de hidrogeno y del solvente empleado. Se obtuvo una alta selectividad hacía el alcohol furfurilico. Con las velocidades iníciales de reacción se determino el orden de reacción global. Los datos experimentales pueden también explicarse usando un modelo Langmuir-Hinshelwood considerando un solo tipo de sitio activo con adsorción disociativa de hidrogeno, siendo la reacción superficial la etapa limitante de la reacción, este modelo se ajusta a los datos experimentales.
PALABRAS CLAVE: Furfural, hidrogenación, efecto SMSI, estudio cinético.
1. INTRODUCTION
Hydrogenation reactions of a, b-unsaturated aldehydes to their corresponding unsaturated alcohol are an interesting type of reaction in fine chemistry. They posses a C=O bond conjugated with a C=C bond. The aim is to hydrogenate the carbonyl group, keeping intact the olefinic function, in spite of the C=C double bond is easily hydrogenated over most conventional catalysts to give saturated aldehydes as the primary products [1-2].
One type of these reactions is the hydrogenation of furfural to obtain furfuryl alcohol which find a variety of applications in chemical industry, as starting material for the manufacture of resins, tetrahydrofurfuryl alcohol (THFA) and as intermediate to obtain lysine, vitamin C, lubricant, dispersing agent and plastisizer [3-4]. Furfuryl alcohol is industrially prepared by the catalytic reduction of furfuraldehyde using Ni and Cu/CrO catalysts. The disadvantage of this type of catalysts is their high toxicity, which causes severe environmental pollution. To design catalysts without chromium for the hydrogenation of furfural with high activity and selectivity is a hard but important task [4].
Liquid phase selective hydrogenation of furfural to produce furfuryl alcohol over gas phase Raney nickel catalysts modified by the impregnation of salts of heteropolyacids have been reported by Liu et al. [5]. Amorphous Ni alloys as Cu/C catalysts have also been used [6] as well as Cu/MgO catalysts [4,7] and mixed Cu-Zn oxides doped with Al, Mn and Fe [4]. Ag/SiO2 catalysts prepared by sol-gel method and Rh-Sn/SiO2 showed selectivities to furfuryl alcohol close to 79% and 93%, respectively [8].
Langmuir-Hinshelwood models involving a bimolecular surface reaction as the rate-determining step have been used to describe the kinetics of furfural hydrogenation over Cu supported on different carbon supports and the results indicated that the catalytic activity are not significantly affected by the nature of the support. However, in those copper catalysts, the metallic component exist as a mixture of Cu0 and Cu(I), and both type of sites may be involved in the catalytic cycle. Reaction order respect to furfural was close to zero on these catalysts whereas the dependence respect to hydrogen was in the range 0.6 to 0.8 [6].
Liquid phase furfural hydrogenation has been studied by Merat et al., [9] over Pt supported catalysts under mild operating condition and they reported that no reaction takes place at 50 °C. Only a few studies on Pt-based catalysts have been published [9-10], and especially Ru and Pd, also seem to be promising [6]. However, fewer results on iridium catalysts has been reported, even though this metal displays interesting hydrogenation ability. Thus, Reyes, et al [11-12] has showed that Ir catalysts displayed a high activity and selectivity in the hydrogenation of C=O group of a, b-unsaturated aldehydes. Recently our group reported the kinetic of the furfural over Ir/Nb2O5 catalysts [12]. In the present work is studied the kinetics of the liquid hydrogenation of furfural over Ir supported on TiO2 catalysts. Additionally, a possible reaction mechanism is proposed.
2. EXPERIMENTAL
TiO2 (Degussa P-25 SBET= 50 m2g-1) was impregnated with H2IrCl6 to obtain Ir/TiO2 catalysts with Ir loading of 1 wt %. The impregnated solid was dried at 343 K for 6 h, calcined in air at 673 K for 4 h and reduced at 773 K (HTR: High temperature reduction) for 2 h.
The BET-surface area was evaluated from nitrogen adsorption isotherms at 77 K and the number of active sites of each catalyst were obtained by hydrogen chemisorption at 298 K, both performed in a Micromeritic ASAP 2010. The surface acidity was determined by temperature programmed desorption, TPD, of NH3. Transmission electronic microscopy (TEM) studies were carried out in a JEOL Model JEM-1200 EXII microscope.
Furfuraldehyde and furfuryl alcohol used in all experiments, both of analytical reagent grade were supplied by Aldrich. Catalytic reactions were carried out in a stainless steel batch reactor at a constant stirring rate (1000 rpm). To carry out the kinetic study over the catalysts only one variable was modified in each experiment, keeping constant all the others. The effect of furfural concentration was studied in the concentration range 0.025 to 0.1 M. The hydrogen partial pressure was studied in the range 0.48 MPa to 0.84 MPa. The temperature was varied in the range 323 to 363 K and the catalyst weight, ranged from 0.1 to 0.3 g.
Prior the experiment, all catalysts were treated in situ under hydrogen flow of 20 cm3min-1 at atmospheric pressure and temperature of 363 K to remove possible surface oxide species generated during handing. The avoid the presence of oxygen, once the reactor was loaded with the reactant mixture and catalyst, the system was flushed with He at atmospheric pressure during 30 min.
Analysis of the reaction mixture and products was carried out using a gas chromatograph Varian 3400 furnished with a HP Wax column of 30 m length and 0.53 mm ID. The GC analysis was performed using a flame ionization detector, using He as carrier. The products of reaction were analyzed in a Varian 3800-Saturn 2000 GC-MS provided with a ionic trap and using the same separation conditions.
Starting with the fresh catalyst, the same catalyst was reused to study possible catalyst deactivation during catalytic test. In all experiments, reactant and product concentrations were measured at different time intervals. From these data, initial rates were calculated allowing obtaining the reaction orders with respect to furfural concentration and H2 pressure.
3. RESULTS AND DISCUSSION
3.1 Catalyst characterization
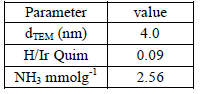
The results of catalysts characterization have been provided previously [11], however some relevant aspects are considered here. These solids when they are reduced at high temperature in H2, lead to a partial reduction of the support, TiO2-x which covers the metal surface, by the so called SMSI effect (strong metal support interaction). This behavior is associated with a decrease of H/Ir ratio in chemisorption measurements (table 1) but without any effect in the metal particle size because there are no changes in the metal particle size in catalysts of Ir/TiO2 reduced at different temperatures of reduction. This phenomenon has been widely studied by TEM [1,11]. Surface acidity measurements were carried out by DTP of ammonia, previously adsorbed at 393 K. Ir/TiO2 exhibits a very low acidity compared with others supports [12].
Table 1. Characterization results of Ir/TiO2 catalyst
3.2 Mass transfer
In the liquid phase hydrogenation of furfuraldehyde, mass-transfer processes (gas-liquid, liquid-solid, and intraparticle diffusion) can influence the rates of reaction [13]. For the kinetic study these diffusional resistances should be absent. The liquid-side mass-transfer coefficient and liquid-solid mass-transfer coefficient depend on the intensity of turbulence in the liquid phase. For this reason highest stirring speed were used (1000 rpm). For prevent intraparticle mass-transfer resistance, it was used smallest catalyst particles sizes (100 mm). An experimental approach following the guidelines of Satterfield and Sherwood (eq. 1) [14] for corroborate the absence of gas-liquid or liquid-solid mass transfer resistance was applied. The equation 1 describes that the reciprocal of the conversion rate as a function of the reciprocal of catalyst mass should give a straight line indicating absence of external mass-transfer limitation.
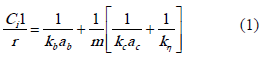
Where the intercept 1/kbab represents a resistance to gas absorption. The term 1/kη represents a resistance associated with the surface reaction, while the term 1/kcac is associated with the transport of hydrogen through the bulk liquid.
The figure 1 displays the results obtained applying this equation. It can be observed the absence of external mass-transfer limitation. Therefore, the mass-transfer rates thus obtained were considerably higher than the maximum reaction rates observed in the kinetic experiments, consequently, it was concluded that external mass transfer did not have an effect on the observed rates in the hydrogenation experiments.
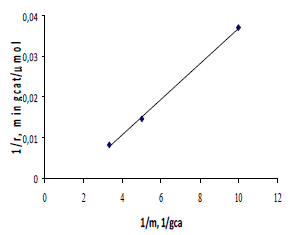
Figure 1. Effect of catalysts weight on the initial rates in the furfural hydrogenation at 363 K, 0.1 M and 0.62 Mpa
3.3 Effect of solvent
Some solvents were used for furfural hydrogenation, such as, ethanol, heptane, ethanol-heptane (1:1). Table 2 summarizes the solvents used, the conversion level at 60 min of reaction time as well as the initial activity expressed as TOF (turnover frequency) for Ir/TiO2 catalysts.
Table 2. Catalytic activity in furfural hydrogenation at 363 K and 0.62 MPa, expressed as conversion, TOF and selectivity at 60 min, SFOL=Selectivity to furfuryl alcohol, SFDA to 2-furaldehyde-diethylacetal
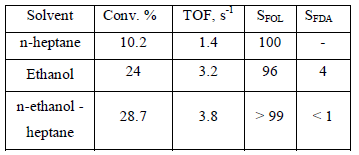
As polarity increases also conversion as well as in the TOF is observed at higher solvent, but the selectivity to the unsaturated alcohol decreases at higher polarity. Similar trends have been reported for other hydrogenation reactions [15]. It is interesting note the synergestic effect of mixed solvents which increases both activity and selectivity.
The results in table 2 indicate clearly that the main product is furfuryl alcohol produced by hydrogenation of C=O bond in the furfural.
The production of furfuryl alcohol is associated with new actives sites in which the metallic component exhibits a partial decoration, with the creation of Ird+ species, which are more active in the polarization of the C=O bond [11,12]. With n-heptane as solvent the selectivity to furfuryl alcohol is 100 %, but acetals are produced with alcoholic solvents, in this case, 2-furaldehyde-diethylacetal (m/z= 39, 97, 125 m/z).
The acetalization takes place over acid sites, specially Brönsted acid sites on the catalyst surface, these sites are placed near to iridium crystals. However, it has been suggested that the acid sites in the catalysts did not modify the active metal properties [16] and favors the electron transfer from acid sites in the support to the active metal sites, by suppressing the hydrogenation of C=C bond. The rate of formation of acetals is faster during the initial period of reaction but the selectivity of acetal decreased with the consumption of reactant and became lower than 1 % at high conversion (figure 2).
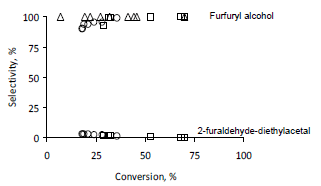
Figure 2. Conversion vs selectivity, effect of solvent in furfural hydrogenation for Ir/TiO2 at 363 K and 0.62 MPa with different solvents used; (Δ) heptane, (○) ethanol, (□) ethanol-heptane 1:1
The Figure 3 shows the hydrogenation products of furfural over Ir/TiO2, arising from the reduction of the C=O group and the formation of 2-furaldehyde-diethylacetal. In this catalyst was not observed the hydrogenation of the furan ring, or other compounds derived from secondary reactions, such as hydrogenolysis of the C-O bond, decarbonylation, hydrogenation and furan ring opening that may appear [17].
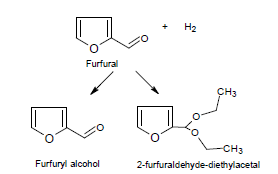
Figure 3. Reaction pathway for furfural hydrogenation over Ir/TiO2
3.4 Analysis of initial rate data
The effect of initial concentration of furfural and H2 pressure on the initial rate data of furfural hydrogenation was studied. Experiments were conducted at various initial concentrations of furfural in the range of 0.025 M to 0.1 M, at 363 K and 0.62 MPa hydrogen partial pressure. The initial rates were expressed as TOF. The order calculated taking out the plot of -log initial concentration vs. -log TOF for Ir/TiO2 was of 0.5 (figure 4).
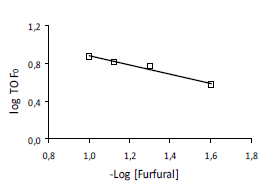
Figure 4. Effect of initial concentration on hydrogenation of furfural over Ir/TiO2 as a plot of -log C0 furfural vs -log TOF.
An explanation for this result can be the adsorption mode of furfural on the catalysts surface. Furfural possibly adsorbs preferentially towards C=C with plane geometry competitive with atop geometry similar in cinnamaldehyde [18]. This could be attributed to the fact that C=C on the ring of furfural are conjugated. At first time reactions the coverage of surface of molecules of furfural with atop geometry is larger, but whiles the reaction progress the surface is totally coverage of furfural in plane geometry.
The effect of the hydrogen partial pressure on the initial rates was studied in the range of 0.48 MPa to 0.84 MPa at constant initial concentration (0.1 M) and temperature (363 K). The results are displayed in figure 5. The order calculated for initial rate was -1,0 using the initial rates expressed as TOF.
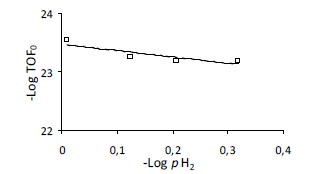
Figure 5. Effect of hydrogen partial pressure in furfural hydrogenation over Ir/TiO2 catalysts as a plot of -log PH2 vs -log TOF
3.5 Kinetic model
Two types of kinetic equations were employed in the quantitative description of the experimental results: first, empirical power-law equations based on initial rates and a second, equations based on the Langmuir-Hinshelwood mechanism. Power-law equations for the hydrogenation of furfural were written as

where i can be Ir/TiO2, p and q are the order obtained at initial rate with respect to furfural concentration and partial pressure of H2. Thus for Ir/TiO2 the global order is -0,5. Because the order with respect to concentration of furfural is fractional, it is desirable to obtain insight into the kinetics via Langmuir-Hinshelwood models.
Considering that the hydrogenation of aldehydes a,b-unsaturated is potentially a complex combination of series and parallel reactions, can be expected a parallel reactions that involve the hydrogenation of both functional groups of the furfural (C=C, C=O), however the results showed for Ir/TiO2 in furfural hydrogenation produces a highly selectivity towards furfuryl alcohol, being the result of the presence of SMSI effect on this type of support. For this catalyst studied the selectivity towards unsaturated alcohol is greater of 98% which can be interpreted as the presence of a single site present (i.e. Ird+ species are preferential).
A typical Langmuir-Hinshelwood model (LH) of a single site can be developed to describe the hydrogenation of furfural towards furfuryl alcohol based on the following assumptions:
- The adsorption of hydrogen occurs in form dissociative and competitive with the organic molecules, due to high H2 partial pressure employed and the hydrogenation surface reaction is irreversible.
- Surface reaction between dissociatively adsorbed hydrogen and adsorbed liquid phase components on the same site is considered as the rate determining step (RDS) while that the adsorption and desorption are in quasi-equilibrium (QE).
- The diffusionals steps not are taken in account.
Considering these observations and assuming on basis of the experimental observation the sequence of reaction is expressed as:
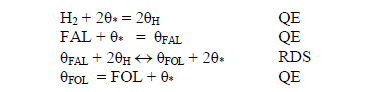
Where q is the active site, FAL is furfural, qH, qFAL, qFOL, q* are respectively the adsorbed hydrogen, furfural adsorbed, furfuryl alcohol adsorbed and vacant sites. Based on the basis asumptions mentioned above, the reaction rate can be written as:
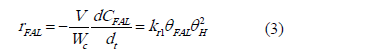
Where V/Wc is the ratio of he reaction volume to the mass of catalyst; CFAL is the concentration of furfural, kr1 is the rate constant for the reaction.
The quasi-equilibrium adsorption /desorption process (steps 1, 2 and 4) provide:
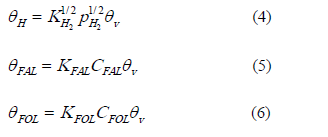
KFAL, KFOL and KH2 are equilibrium adsorption constants. These expressions can be substituted into the RDS step.
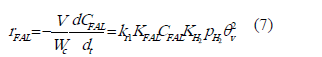
Considering that the expression for the fractional surface coverage of sites is:
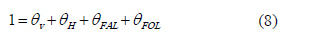
In agreement of the results of hydrogenation reactions at 363 K with addition of the unsaturated alcohol (furfuryl alcohol) to the reaction mixture in the beginning had no effect on the rate of reaction the product term qFOL was omitted.
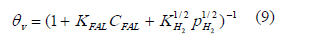
Finally the reaction rate for model 1 is expressed as:
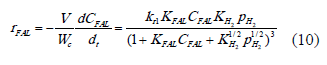
A second LH model was proposed using the assumptions presented for model I, except for assumption 2, which assumes that addition of hydrogen takes place in two steps and is considered that the addition of first hydrogen is the RDS. Thus the sequence LH can be expressed as:
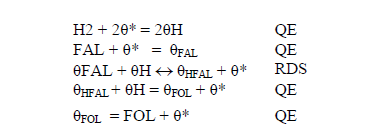
The reaction rate can be written as:
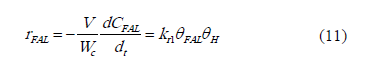
With the same procedure describe above, the rate expression for model II can be expressed as:
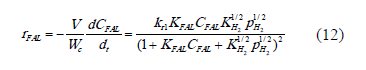
3.6 Confirmation of reaction mechanism
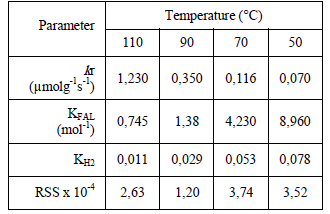
In order to select a suitable rate equation, a nonlinear least-squares regression analysis was used for each rate equation to obtain the best values of the parameters using the values of the rate constants were fitted of the experimental concentration versus time profiles of the chemical species involved in the reaction, using a non-linear regression program with an iterative Gauss-Newton method. The objective function to be minimized is residual sum of squares, RSS=∑(r*-rcalc)2 where r* is the experimental relative rate, rcalc is the relative concentration calculated with the model. A comparison of the RSS between the models described above, indicates that model II gives the best fit to the experimental data (table 3).
Table 3. Optimized rate parameters for the hydrogenation of furfural at different temperatures, RSS between experimental data and results simulated by mechanism II
The comparison of the experimental concentrations and calculated concentration shows good agreement between the model predictions and the model observed values (figure 6).
Figure 5. Parity plot of calculated concentrations (Eq. 12) with experimental concentrations at various temperatures
The reactions were carried out at different temperatures in the range of 323 to 363 K. Table 3 displays the optimized rate parameters at different temperatures for model II. From the temperature dependence of rate parameters the activation energy was calculated. The surface activation energy near of 12 kcal mol-1 is similar that the reported for Vaidya et al, for furfural hydrogenation over Pt/C [13]. The enthalpy of adsorption and the entropy of adsorption of furfural and hydrogen were calculated from KFAL and KH2 using the van't Hoff equation, these values are listed in Table 4. Both enthalpies and entropies of adsorption are negative, as they should be, and quantitatively satisfies guidelines established, according to their thermodynamic consistency [19].
Table 4. Thermodynamic parameters for adsorption KR from expressions encountered for furfural hydrogenation over Ir/TiO2

4. CONCLUSIONS
Furfural hydrogenation over Ir/TiO2 catalysts reduced to 773 K does not present limitations of mass transfer to describe a kinetic inadequate behavior. The solvent affects the selectivity of the reaction due to reactions of acetalization producing 2-furaldehyde-diethylacetal. Initial rates describes that Ir/TiO2 posses a -0,5 order global. A kinetic model Langmuir-Hinshelwood of a single-site that involves dissociative adsorption of first hydrogen on the organic molecule as the rate-controlling step provided the best fit of the experimental data. Thus thermodynamic parameters obtained for this model demonstrates that the mechanism proposed is consistent physically and it describes the high selectivity towards the unsaturated alcohol due principally to SMSI effect.
NOTATION
ab interfacial area at gas-liquid interface
ac specific surface area of catalyst
kb mass transfer coefficient for gas absorption across gas-liquid interface
kc mass transfer coefficient for hydrogen transport through the bulk liquid
q active site.
qv vacant active site.
h catalyst effectiveness factor
CFAL bulk liquid phase concentration of furfural
CFAL-H half-hydrogenated intermediate
k apparent rate constant
KFAL adsorption equilibrium constant for single site adsorption of furfural
KH2 adsorption equilibrium constant for hydrogen
KFOL adsorption equilibrium constant for furfuryl alcohol
m catalyst loading (density), g cat/L solution
p, q Orders reaction
r rate of furfural hydrogenation, mol/L min
pH2 gas phase partial pressure of hydrogen
rFAL specific rate of furfural hydrogenation, µmol g-1s-1
wc catalyst mass
RSS Residual sum of squares
ACKNOWLEDGEMENTS
We thank to COLCIENCIAS-SENA and DIN-UPTC for the financial support under the project Nº 110948925094.
REFERENCES
[1] GALLEZOT, P., RICHARD, R. Selective hydrogenation of a,ß-unsaturated aldehydes. Catal. Rev. Sci. Eng, 40, 81-126, 1998. [ Links ]
[2] ARVELA, M., HÁJEK, J., SALMI, T., MURZIN, D.Y. Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts, Appl. Catal. A., 292, 1-59, 2005. [ Links ]
[3] McKETTA, J.J., CUNNINGHAM, W.A. Encyclopedia of Chemical Processing and Design; Marcel Dekker: New York, Vol. 24, pp. 63, 1986. [ Links ]
[4] NAGARAJA, B.M., SIVA KUMAR, V., SHASIKALA, V., PADMASRI. A.H., SREEDHAR. B. A highly efficient Cu/MgO catalyst for vapour phase hydrogenation of furfural to furfuryl alcohol, Catal. Comm., 4, 287- 293, 2003. [ Links ]
[5] LIU, B.J., LU, L.H., WANG, B.C., CAI T.X., KATSUYOSHI, I. Liquid phase selective hydrogenation of furfural on raney nickel modified by impregnation of salts of heteropolyacids. Appl. Catal. A. 171, 117, 1997. [ Links ]
[6] RAO, R., BAKER, R.T., VANNICE, M. Furfural hydrogenation over carbon supported copper. Catal. Lett. 60, 51-57, 1999. [ Links ]
[7] NAGARAJA, B.M., PADMASRI A.H., Raju, B.D., Rao, K.S. Vapor phase selective hydrogenation of furfural to furfuryl alcohol over Cu-MgO coprecipitated catalysts. J. Mol. Catal. A: Chem. 265, 90-97, 2007. [ Links ]
[8] CLAUSS, P. Selective hydrogenation of a,ß-unsaturated aldehydes and other C=O and C=C bonds containing compounds. Topics Catal., 5, 51, 1998. [ Links ]
[9] MERAT, N., GODAWA, C., GASET, A.J. High selective production of tetrahydrofurfuryl alcohol: Catalytic hydrogenation of furfural and furfuryl alcohol. Chem. Technol. Biotechnol. 48, 145-159, 1990. [ Links ]
[10] KIJENSKI, J., WINIAREK, P., PARYJCZAK, T., LEWICKI, A., MIKOLAJSKA A. Platinum deposited on monolayer supports in selective hydrogenation of furfural to furfuryl alcohol. Appl. Catal. A. 233, 171-182, 2002. [ Links ]
[11] REYES, P., ROJAS, H., PECCHI, G., FIERRO, J.L.G. Liquid-phase hydrogenation of citral over Ir supported catalysts. J. Mol. Catal. A: Chem. 179, 293-299, 2002. [ Links ]
[12] ROJAS, H., BORDA, G., MARTÍNEZ J.J, ROSAS, D., REYES P. Hidrogenación de furfural sobre catalizadores Ir/Nb2O5. Estudio Cinético. Dyna 155, 115-122, 2008. [ Links ]
[13] VAIDYA. P., MAHAJANI. V.V. Kinetics of liquid phase hydrogenation of furfuraldehyde to furfuryl alcohol over a Pt/C catalyst. Ind. Eng. Chem. Res., 42, 3881-3885, 2003. [ Links ]
[14] SATERFIELD, C.N., SHERWOOD T.K. The role of diffusion in catalysis. Addison'wesley, Reading M.A. pp 43-55. 1963. [ Links ]
[15] REYES, P., ROJAS, H. The solvent effect in the hydrogenation of citral over Ir and Ir-Fe/TiO2 catalysts. J. Chil. Chem. Soc, 52, 1155-1159, 2007. [ Links ]
[16] YILMAZ, S., UCAR, S., ARTOK, L., GULEC, H. The kinetics of citral hydrogenation over Pd supported on clinoplite rich natural zeolite. Appl. Catal. A 287, 261-266, 2005. [ Links ]
[17] MERLO, A.B., VETERE, V., RUGGERA, J.F., CASELLA, M.L. Bimetallic PtSn catalyst for the selective hydrogenation of furfural to furfuryl alcohol in liquid-phase. Catal. Comm., 10, 1665-1669, 2009. [ Links ]
[18] BREEN. J.P., BURCH. R., GÓMEZ-LÓPEZ J., GRIFFIN. K., HAYES. M. Steric effects in the selective hydrogenation of cinnamaldehyde to cinnmayl alcohol using an Ir/C catalyst. Appl. Catal A., 268, 267-273, 2004. [ Links ]
[19] VANNICE. M.A., HYUN. S.H., KALPAKCI. B. LIAUH. W.C. Entropies of adsorption in heterogeneous catalytic reactions. J. Catal., 56, 358-364, 1979. [ Links ]

