










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkDYNA
versão impressa ISSN 0012-7353
Dyna rev.fac.nac.minas vol.81 no.187 Medellín set-/out. 2014
https://doi.org/10.15446/dyna.v81n186.46092
http://dx.doi.org/10.15446/dyna.v81n187.46092
The importance of being chemical affinity. Part V: The fruits
La importancia de llamarse afinidad química. Parte V: Los frutos
Guillermo Salas-Banuet a, José Ramírez-Vieyra b, Oscar Restrepo-Baena c, María Noguez-Amaya d & Bryan Cockrell e
a Universidad Nacional Autónoma de México, México, salasb@unam.mx
b Universidad Nacional Autónoma de México, México, jgrv@unam.mx
c Universidad Nacional de Colombia, Colombia, ojrestre@unal.edu.co
d Universidad Nacional Autónoma de México, México, nogueza@unam.mx
e University of California, Berkeley, USA, bryan.cockrell@berkeley.edu
Received: December 10th, 2013. Received in revised form: May 7th, 2014. Accepted: June 13th, 2014
Abstract
Through the meticulousness in experimentation, the incorporation of discoveries in physics and the application of the logic of algebra to make chemistry's language systematic and universal, the level of thought achieved in chemistry during 18th century gave a foundation to the discipline that would allow it to evolve and transform. The discovery, comprehension and application of electricity in the 19th century supplied chemistry with analogous units, which in turn, facilitated increased understanding of the field and permitted the proposal of new theories. Developments in thermodynamics allowed for the presentation of qualitative scales of affinity and electronegativity. The discovery of the atom and electron and the emergence of quantum physics restructured chemical thought, permitting for the establishment, in the first half of the 20th century, of the first quantitative scale of electronegativity, which offered chemistry a new dynamism, facilitating the proposal of other scales, based on diverse approaches.
Keywords: Chemical Affinity, science, electroaffinity, electronegativity, thought, history.
Resumen
El nivel de pensamiento alcanzado en la química durante el siglo XVIII, a través de la exactitud en la experimentación, la asimilación de los descubrimientos de la física y la aplicación de la lógica del álgebra para hacer del lenguaje químico uno sistemático y universal, permitió consolidar una base para que la química evolucionara y se revolucionara. El hallazgo, entendimiento y aplicación de la electricidad en el siglo XIX, le proveyó de elementos analógicos para su mejor comprensión y para la propuesta de nuevas teorías; la termodinámica le permitió presentar escalas cualitativas de afinidad o electronegatividad. El descubrimiento del átomo, del electrón y la física cuántica redimensionaron el pensamiento químico, permitiendo el planteamiento, en la primera mitad del siglo XX, de la primera escala cuantitativa de electronegatividad, la cual inyectó un nuevo dinamismo a la química, induciendo la propuesta de otras escalas, basadas en diversas racionalizaciones.
Palabras clave: Afinidad química, ciencia, electroafinidad, electronegatividad, pensamiento, historia.
1. Introducción
Existe una anécdota relativa a la muerte del químico, biólogo y economista parisino Antoine Lavoisier (1743-1794), que bien puede ser una leyenda: el 7 de mayo de 1794, día anterior a morir guillotinado durante la revolución francesa, con la finalidad de dar alguna utilidad científica a su muerte, concertó con algunos colegas que intentaría mantenerse parpadeando después de que la guillotina le cortase la cabeza para demostrar que la muerte no era inmediata; parpadeó 12 veces. Si esta historia es cierta, muestra la naturaleza profundamente científica de Lavoisier y de los pares de su época.
Se ha considerado que el nacimiento de la ciencia moderna se sitúa en un periodo de ciento cuarenta y cuatro años, comprendido entre 1543 -año de la publicación del libro "Sobre las revoluciones de las esferas celestes" del astrónomo, abogado y médico polaco Nicolás Copérnico (1473-1743)- y 1687 -cuando se publicó "Principia Mathematica"-, del físico, filósofo, teólogo, inventor, alquimista y matemático inglés Isaac Newton (1642-1727). También se ha dicho que los cambios que tuvieron lugar durante ese periodo -la concepción heliocéntrica del sistema solar, la tipificación de la gravitación universal, la comprensión de la luz, el vacío, los gases, el cuerpo y la vida microscópica- determinaron la llamada Revolución científica porque transformaron total e irremediablemente la concepción que se tenía de la naturaleza; con ella se desecharon la noción del mundo aristotélico y la reflexión aleatoria y sobrenatural de la Edad Media, las cuales fueron sustituidas por la racionalidad sobria, matemática y acumulativa de la Edad Moderna. Pero Newton también fue un alquimista que buscaba la piedra filosofal, que estudiaba la cronología de la Biblia convencido de que le permitiría prevenir el apocalipsis que vendría y las leyes de la acción divina en la naturaleza para demostrar la existencia y providencia de Dios; puede considerarse a Newton un cuasi-místico hechizado por los rosacruces, la astrología y la numerología, quien creía que Moisés conocía la teoría heliocéntrica de Copérnico y su propia teoría de la gravedad; y un esforzado investigador de la forma exacta del Templo de Salomón, al que consideraba la mejor guía para descubrir la topografía de los cielos [1,2]. Sin embargo, hay estudios que sugieren que los descubrimientos científicos de Newton no podrían haber sido realizados si no ha sido por sus investigaciones alquímicas [3, 4]. Así, hablar de una revolución científica en un periodo de tiempo tan reducido parece poco sustentable y, tal vez, habría que ampliarlo hasta el siglo XVIII, abarcando a los cambios que dieron origen a la química moderna y que cristalizaron con Lavoisier [5]. El descubrimiento de la electricidad y la experimentación a la que estuvo sujeta, permitieron que los químicos utilizaran sus conceptos para reinterpretar lo que se sabía de la afinidad entre elementos y sustancias y comprendieran mejor lo que sucedía durante su unión y separación. Con ello y a lo largo de muchos años, el concepto de afinidad dejó de usarse para ser sustituido directamente por el de electronegatividad, sin dejar de significar lo mismo: Tendencia de un átomo o molécula a reaccionar o combinarse con átomos o moléculas de diferente constitución química [6], la propensión que existe entre átomos o moléculas diferentes a unirse para formar una sustancia o compuesto o, en sentido inverso, la resistencia que muestran los átomos o moléculas de las sustancias o compuestos a ser separados.
2. Antecedentes
En esta serie de escritos [5, 7-9] se ha plasmado una idea de la historia occidental acerca del esquivo concepto de la afinidad química. Históricamente, en Mesopotamia (antigua Irak y noreste de Siria), Egipto y Persia (ahora Irán), los hombres que manipulaban constantemente los materiales, entendieron que podían cambiarlos usando altas temperaturas; el primer pensamiento que utilizaron para explicar esas transmutaciones fue el religioso. Sin embargo, en cualquier época, siempre han existido personas que no se han apoyado en el pensamiento religioso para explicar el mundo; personas creativas, observadoras, perfeccionistas y reflexivas; unas involucradas en los procesos de producción de materiales artificiales, que pudieron entender, mejorar y crear; y otras dedicadas a pensar sistemáticamente sobre el mundo, interpretándolo. Son estas quienes, de manera inconsciente, comenzaron a desarrollar la idea de que algo en la materia podía separarse y unirse para ser cambiada. Es en Grecia, primero en Jonia, luego en Atenas y por último en Alejandría-entre los siglos VI a.C. y II d.C.- que se utilizó sistemáticamente un pensamiento independiente del religioso, llamado aristotélico, para explicar al mundo. Allí nacieron los conceptos básicos necesarios para construir el concepto de afinidad química; fue al final de ese periodo que nació la alquimia. Más tarde en Europa, durante aproximadamente un milenio (la Edad Media), con el rígido control que ejerció la Iglesia Cristiana de Roma sobre el pensamiento, todo se interpretó usando un pensamiento religioso colectivo, basado en los conceptos de esa religión. Paralelamente floreció la alquimia, produciendo una base para la química práctica. Es en el siglo IX d.C., que uno de los fieles de la iglesia cristiana inició la defensa del pensamiento individual que, junto con el descubrimiento de los escritos aristotélicos, promovería el desarrollo del conocimiento basado en un pensamiento independiente del religioso que, respecto a la afinidad química, daría sus primeros frutos durante el siglo XVII.
3. Edad moderna. Electricidad, temodinámica y estructura atómica en la afinidad química
Claude Louis Berthollet (1748-1822), médico y químico francés, quien sería uno de los colaboradores más importantes de Lavoisier, formó parte del grupo de científicos que en 1798 acompañaron a Napoleón a Egipto. Allí, Berthollet se interesó por los lagos Natron, palabra griega para la sosa o carbonato de sodio, Na2CO3×nH2O, los cuales se encuentran en una gran depresión al oeste de El Cairo. Al visitarlos, efectuó análisis químicos que indicaban que el agua de los lagos contenía nitrato de sodio, NaNO3, carbonato de sodio, Na2CO3 y una pequeña cantidad de sulfato de sodio, NaSO4, en diversas proporciones. Las observaciones que realizó sobre los productos sólidos formados (cloruro de sodio, NaCl, carbonato de sodio y salitre, NaNO3) en las varias condiciones en que los encontró, lo llevaron a razonar y concluir que condiciones tales como la temperatura, la concentración relativa, las cantidades de los reactantes o sus estados de agregación, afectaban la naturaleza y dirección de las afinidades en una reacción química. Esto lo llevó a concebir un nuevo sistema en química que desarrolló en su libro Essai de Statique Chimie [10], publicado en 1803, en el que estableció un nuevo concepto de equilibrio químico, rompiendo la visión de la afinidad que cumple con leyes estrictas e invariantes.
En 1795, Alessandro Volta (1745-1827), profesor italiano de física de la Universidad de Padua, demostró que la electricidad podía producirse colocando dos piezas de metales diferentes con un líquido o un paño húmedo entre ellas, con lo que creó la primera batería de corriente eléctrica -muy cara-, comenzándose a comprender sus propiedades básicas. En 1802, el químico británico Humphry Davy (1778-1829) -utilizando una pila enorme- consiguió aislar, además del magnesio, bario, estroncio, calcio, y boro, dos nuevos metales mucho más baratos, el sodio y el potasio, con lo cual la electricidad comenzó a ser sujeto de investigación importante. Pocos años después (1807) Davy pudo sugerir, en la publicación On some chemical agencies of electricity [11] que la afinidad química provenía de las energías eléctricas de los cuerpos. Enunció que, si dos tipos de partículas con cargas muy opuestas se encontraban, éstas se unirían tan fuertemente que formarían un compuesto estable y, basado en su observación de que durante las neutralizaciones química y eléctrica se producían distintas intensidades de calor, explicó que la afinidad entre los cuerpos se podía modificar, cambiando sus energías eléctricas al utilizar una pila voltaica o al calentarlos. Expuso que era posible medir experimentalmente la energía eléctrica necesaria para descomponer una sustancia o para establecer su carácter positivo o negativo y propuso una escala de afinidad química relativa. También señaló que había una relación entre la acidez y la electricidad negativa y entre la alcalinidad y la electricidad positiva, con la que se podría especificar la posición de cada sustancia en esa escala. Quien aplicó en la química esta comparación entre ácido-alcalino y electricidad negativa-positiva fue el físico y químico italiano Amedeo Avogadro (1776-1856), al observar las inconsistencias de la definición de acidez absoluta de Lavoisier y tomar en cuenta los puntos de vista de Berthollet sobre la acidez y la afinidad. En su trabajo Idées sur l'acidité et l'alcalinité [12], de 1809, Avogadro sugirió que los conceptos de acidez y alcalinidad deberían abarcar todas las interacciones químicas. Propuso que cada sustancia simple tiene una acidez o alcalinidad relativa y que ésta se puede ubicar en una escala universal de acidez. Además, situó en primer lugar de su escala al oxígeno, acuñando el término "oxigenidad", para sustituir al de acidez, que pasaría a describir los valores de la oxigenidad relativa entre dos sustancias: a mayor separación entre dos sustancias en la escala, mayor antagonismo habría entre ellas y más grande sería su afinidad química. Sin embargo, Berthollet [10] señaló que la oxigenidad de un material sólo se manifestaba para algunos estados de agregación. Así, la escala de oxigenidad mostraría la habilidad de una sustancia para adquirir una electrificación negativa, equivalente a una escala simple de electronegatividad. Al correlacionar la oxigenidad con el calor específico de varias sustancias -al que consideraba como medida de la afinidad de las sustancias por el calor- Avogadro encontró una relación de la oxigenidad con los volúmenes atómicos relativos de los elementos, concluyendo que los elementos fuertemente oxigénicos tenían volúmenes atómicos grandes, el equivalente de la relación que existe entre la electronegatividad y el tamaño de los átomos de los elementos.
El sueco Jöns Berzelius (1779-1848), contemporáneo de Avogadro, propuso una teoría electroquímica para la afinidad química. En 1825, en su libro Lärbok i kemien, traducido y publicado en alemán como Lehrbuch der Chemie [13], situó a las sustancias simples en una escala similar a la de oxigenidad, a la que llamó de electronegatividad, donde la oposición ácido-alcalino tenía su equivalente en el antagonismo electronegativo-electropositivo. Berzelius incluyó en su escala a los 54 elementos conocidos en su época, separando con una línea a las especies eminentemente electronegativas de las predominantemente electropositivas. Lo realmente novedoso en su teoría residía en la explicación de que la cantidad de calor producido -o la cantidad electricidad neutralizada- en una reacción, era una medida de la afinidad química entre los reactivos. Sugirió también que al calentarlos aumentaba su carga eléctrica -lo cual explicaba la aceleración de la reacción- y que el calor producido se debía a la destrucción de sus cargas durante la formación del producto [14].
Un hecho fundamental fue la racionalización, inducida por el empirismo, durante el estudio de las propiedades físicas de la atmósfera y otros gases, que llevó al británico John Dalton (1766-1844) al planteamiento de su teoría atómica. En su tabla de pesos atómicos relativos -la primera en publicarse- Dalton presenta seis elementos: hidrógeno, oxígeno, nitrógeno, carbono, azufre y fósforo, asignándole al átomo de hidrógeno un peso unitario. Interesado en la elasticidad de los fluidos, advirtió que bajo diferentes presiones, tomando en cuenta el principio de conservación de la materia, los gases del mismo peso debían de tener configuraciones diferentes. Al crear nuevos gases y determinar sus pesos pudo introducir una nueva nomenclatura, actualmente vigente, v. gr., N2O, NO y NO2; y al estudiar las reacciones químicas de precipitación, en las que al unir dos sustancias líquidas transparentes estas producen un sólido o un gran cambio de color, se convenció que existía una reconfiguración de una entidad básica, los átomos, y que los elementos y compuestos químicos estaban constituidos por ellos. En su libro publicado en 1808, A new system of chemical philosophy [15], Dalton estableció los puntos principales de su teoría, sumarizados en cinco: 1. Los elementos están hechos de partículas diminutas llamadas átomos que son indestructibles e indivisibles; 2. Todos los átomos de un determinado elemento son idénticos; 3. Los átomos de un elemento son diferentes a los de cualquier otro elemento, los átomos de elementos diferentes se pueden distinguir unos de otros por sus respectivos pesos atómicos relativos; 4. Los átomos de un elemento se combinan con los átomos de otros elementos para formar compuestos químicos, un compuesto dado siempre tiene el mismo número relativo de tipos de átomos; 5. Los átomos no se pueden crear ni dividir en partículas más pequeñas, ni se destruyen en el proceso químico. Una reacción química simplemente cambia la forma en que los átomos se agrupan.
Según asegura el norteamericano George F. Barker (1835-1910) en su el libro A Text-book of Elementary Chemistry [16], para 1870 los químicos podían determinar tres propiedades atómicas: 1. El peso atómico, que permitía medir la cantidad de materia presente, calcular la composición de las sustancias y cuantificar las reacciones químicas; 2. La valencia atómica, que evaluaba el poder de combinación de un átomo, permitía prever la estructura química y predecir el número de isómeros posibles para una composición dada; y 3. La electronegatividad atómica, que expresaba el poder de combinación de un átomo.
En aquel entonces se publicaban tablas para cada propiedad. La tabla de las electronegatividades era la publicada por Berzelius en 1836, con nueve elementos adicionales. Barker ilustró la forma en que los químicos usaban la electronegatividad para explicar la reactividad de las moléculas isotípicas o isoestructurales, a través de ácidos, bases y sales. En la nueva teoría, los óxidos de los no metales o átomos electronegativos (v. gr. SO3) correspondían a los ácidos de la teoría dualista de la afinidad; los óxidos de los metales o átomos electropositivos (v. gr. K2O), a las bases y la reacción entre los óxidos básicos y los óxidos ácidos (v. gr. K2OSO3 o K2SO4) producían las sales. Por otra parte, la teoría de tipo había aceptado como válida la teoría de acidez del hidrógeno, que postulaba que al sustituir uno o los dos átomos de hidrógeno en la molécula de agua (H-O-H), por un átomo o radical monovalente con valores de valencia idénticos -para poder preservar la estructura "tipo" del agua- se podían obtener todos los ácidos (R--O-H), bases (R+-O-H) y sales (R--O-R+). En el mismo sentido, Josiah Cooke (1827-1894), utilizó signos negativos y positivos en sus fórmulas químicas para expresar las polaridades relativas y para el oxígeno bivalente usó dos polos de distinto signo (H--+O--K+). En su libro de texto de 1874, The New Chemistry [17], subrayó que la teoría tipo no explicaba las características distintivas de los ácidos, bases y sales, sin recurrir al carácter electroquímico de los átomos contenidos en esos compuestos, a partir del simple tipo del agua; esto significaba que debería existir un continuo de posibles derivados de agua (hidratos), cuyas acideces y alcalinidades aumentaban, respectivamente, desde el centro hacia los extremos de la tabla.
Hacia 1880, los datos sobre los pesos atómicos y las valencias de los elementos ya eran comunes y numerosos. Esto permitió a los químicos encontrar gran cantidad de pautas que derivaron en la propuesta de la ley periódica. Dentro de este nuevo orden, la importancia de la clasificación de las sustancias de acuerdo a su electronegatividad disminuyó considerablemente. Finalmente, Lothar Meyer (1830-1895) señaló que la electronegatividad, como las otras propiedades atómicas, era periódica, uniendo así las dos clasificaciones.
El primer quinto del siglo XX estuvo marcado por dos hechos ocurridos a finales del siglo XIX:
El primero, la publicación del tercer volumen, en 1899, de Lectures on Theoretical and Physical Chemistry [18], de Jacobus van't Hoff (1852-1911), que dio lugar a la búsqueda de relaciones entre la entalpía de reacción y la electronegatividad. Al comparar las entalpías de reacción de cantidades equivalentes de azufre, cloro, iodo, hidrógeno y sodio, van't Hoff se percató de que la mayor facilidad para combinarse -y la máxima generación de calor- se presentaban cuando reaccionaban el sodio y el cloro, elementos con la mayor diferencia de electronegatividad. Robert M. Caven (1870-1934) y George D. Lander presentaron resultados similares en su libro de texto, Systematic Inorganic Chemistry from the Standpoint of the Periodic Law [19], publicado en 1906, indicando la tendencia de que cuanto más diferencias hubiera en el carácter electroquímico de los elementos, mayores serían los valores del calor de reacción. Otto Sackur (1880-1914) también daba cuenta, en su libro de 1913, A Text Book of Thermo-Chemisty and Thermodynamics [20], de que la máxima generación de calor se daba cuando se combinaban los elementos ubicados en los extremos opuestos del sistema periódico y que, al aumentar el peso atómico de los elementos que reaccionaban para formar óxidos, cloruros, sulfuros, etc., el calor de formación generalmente disminuía. También observó que la tendencia a formar iones negativos (la electroafinidad) presentaba el mismo comportamiento y que el potasio y el sodio, elementos con gran tendencia a formar cationes, y el cloro, con gran tendencia a formar aniones, producían calores de formación grandes cuando se combinaban con elementos con carácter eléctrico opuesto.
Y el segundo, el descubrimiento del electrón en 1897 por el físico británico Joseph John Thomson (1856-1940) quien, junto a sus colegas John Sealy Townsend (1868-1957) y Harold Albert Wilson (1874-1964), demostró experimentalmente que los rayos catódicos eran realmente partículas subatómicas y no ondas, átomos o moléculas. Existía ya un antecedente: la explicación sobre la estructura del átomo propuesta por Hermann von Helmholtz (1821-1894) en On the Modern Development of Faraday's Conception of Electricity, de 1881 [21]. En su artículo, von Helmholtz recurre a la teoría dualística de la electricidad para proponer un átomo neutro, constituido por partículas eléctricas en movimiento, positivas y negativas en igual cantidad, asociadas a un núcleo central. Estas partículas podían ser desprendidas para crear cationes o aniones, dependiendo de la habilidad del átomo para ganarlas, perderlas o retenerlas, dándole así su carácter electropositivo o electronegativo.
El descubrimiento del electrón estimuló el pensamiento científico llevando a la propuesta de dos diferentes modelos [22]:
- En su monografía de 1903 Die Diessoziierung und Umwandlung chemischer Atome, Johannes Stark (1874-1957), propuso la tendencia de los átomos a saturarse con electrones negativos, que difería de un elemento a otro de acuerdo con la magnitud de su energía de ionización: a mayor fuerza con la que un átomo mantenía sus electrones, mayor energía de ionización. Entonces sería posible construir una escala de electronegatividad cuantitativa midiendo estas energías; y
- Para el químico inglés Geoffrey Martin (1881-?) sólo un tipo de electrón era responsable de la valencia química. En su libro de 1905. Researches on the Affinity of the Elements and on the causes of the chemical similarity or dissimilarity of elements and compounds afirmaba que los electrones provocaban que un átomo fuera electropositivo o electronegativo, dependiendo de qué tan débil o fuertemente eran retenidos.
En 1911, el alemán Walter Nernst (1864-1941), promotor y congresista en la Primera Conferencia Solvay, en Bruselas, cuyo tema principal fue la Radiación y los Cuantos, presentó un trabajo donde proponía que la afinidad química de los elementos y los radicales eran diferentes, debido a los electrones positivos y negativos; los que tendían a combinarse con electrones positivos formaban grupos de elementos positivos y los que tenían afinidad hacia los electrones negativos producían grupos negativos. Así, dichos modelos fueron propuestos, uno dualístico y el otro unitario, aunque para muchos científicos eran equivalentes. El concepto de un electrón unitario para la interpretación de la electronegatividad ya era bastante aceptado hacia 1913, por lo que el polaco Kasimir Fajans (1887-1957) propuso en la ley de los desplazamientos radiactivos -conocida como ley de Soddy-Fajans- que el carácter electronegativo de un átomo se determinaba como su tendencia para soltar o tomar electrones negativos, sin justificación alguna.
4. La afinidad química durante la segunda revolución científica
Se está viviendo la segunda revolución científica, la cual, según algunos científicos, comenzó a principios del siglo XX con el descubrimiento simultáneo del cuanto, el gen y el inconsciente. En este contexto, la química y su concepto de afinidad encuentran una nueva propuesta en el trabajo de Linus Pauling (1901-1994) quien, junto con Don M. Yost (1893-1977), propuso la primera escala cuantitativa para la electronegatividad (1932).
4.1. La química posmoderna. La electronegatividad de Pauling y la termodinámica
El posmodernismo es una renovación fundamental de las formas tradicionales del pensamiento, el arte, la cultura, la literatura, la filosofía y la vida social y la ciencia. Pauling comprendió el trabajo que sus antecesores de la Edad Moderna habían hecho. Así, planteó una experimentación que lo llevó a proponer una definición de electronegatividad y a materializar una escala cuantitativa para la misma. Primero, Pauling y Yost (1932) propusieron y demostraron experimentalmente su llamado postulado de la aditividad: el valor de la energía de un enlace covalente sencillo entre dos átomos diferentes (enlace heteronuclear), es muy parecido al promedio aritmético de las energías de los enlaces entre los átomos iguales (enlace homonuclear). Luego, Pauling explicó la diferencia entre estos valores con la presencia de un carácter iónico (compartir electrones de manera desigual) en ese enlace covalente, de manera que la energía de éste aumentaría. Cuanto más diferentes fueran los átomos involucrados en el enlace (A y B), mayores serían la energía del enlace y su carácter iónico. Pauling confirmó esto midiendo los calores de formación de varios compuestos y propuso que la diferencia entre esas dos energías (D) podría calcularse como: D = (XA - XB)2; siendo XA y XB las electronegatividades de los átomos A y B, respectivamente. Ya que la diferencia de electronegatividades es proporcional a 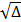 , las unidades de éstas serían raíz cuadrada de la energía [23]. Para Pauling (1932), los valores de D obtenidos sugerían la posibilidad de colocar a los átomos a lo largo de una escala de electronegatividad y supuso que D era una función de la separación lineal de los lugares geométricos de dos átomos en la escala [24]. Pero así sólo se podían definir diferencias de electronegatividad. Para construir la escala se necesitaba establecer un punto de referencia: ya que el hidrógeno forma enlaces covalentes con una gran variedad de elementos, se eligió éste elemento. Tomando el valor de electronegatividad de 2.1 obtenido por Mulliken en 1934 [25], Pauling construyó una escala para 10 elementos (C, N, O, Cl, F, Br, H, I, P y S) [26] que, para 1960, contaba con más de 50 [27]. Al ser los datos termodinámicos más exactos y de más fácil acceso, algunos investigadores -como Allred en 1961 [28] y Fung en 1965 [29]- revisaron los valores de electronegatividad obtenidos por Pauling, dando lugar a los llamados valores de electronegatividad de Pauling revisados, que se usan cotidianamente; el valor de 2.1 para el H, cambió a 2.2. Bergmann y Hinze consideraron que, al tomar el valor de electronegatividad calculado por Mulliken para el H, en 1934, Pauling transformó su escala en absoluta [25]. Para construir su escala de electronegatividades (XP), en 1960, Pauling utilizó conceptos de la teoría del enlace de valencia y, apoyándose en los argumentos de la mecánica cuántica y en los suyos propios, concluyó que los enlaces iónicos y covalentes eran casos extremos y que casi todos los enlaces estaban constituidos por una combinación de ambos. De esta manera, el carácter iónico parcial (el grado de polaridad), I, del enlace se podría calcular como
, las unidades de éstas serían raíz cuadrada de la energía [23]. Para Pauling (1932), los valores de D obtenidos sugerían la posibilidad de colocar a los átomos a lo largo de una escala de electronegatividad y supuso que D era una función de la separación lineal de los lugares geométricos de dos átomos en la escala [24]. Pero así sólo se podían definir diferencias de electronegatividad. Para construir la escala se necesitaba establecer un punto de referencia: ya que el hidrógeno forma enlaces covalentes con una gran variedad de elementos, se eligió éste elemento. Tomando el valor de electronegatividad de 2.1 obtenido por Mulliken en 1934 [25], Pauling construyó una escala para 10 elementos (C, N, O, Cl, F, Br, H, I, P y S) [26] que, para 1960, contaba con más de 50 [27]. Al ser los datos termodinámicos más exactos y de más fácil acceso, algunos investigadores -como Allred en 1961 [28] y Fung en 1965 [29]- revisaron los valores de electronegatividad obtenidos por Pauling, dando lugar a los llamados valores de electronegatividad de Pauling revisados, que se usan cotidianamente; el valor de 2.1 para el H, cambió a 2.2. Bergmann y Hinze consideraron que, al tomar el valor de electronegatividad calculado por Mulliken para el H, en 1934, Pauling transformó su escala en absoluta [25]. Para construir su escala de electronegatividades (XP), en 1960, Pauling utilizó conceptos de la teoría del enlace de valencia y, apoyándose en los argumentos de la mecánica cuántica y en los suyos propios, concluyó que los enlaces iónicos y covalentes eran casos extremos y que casi todos los enlaces estaban constituidos por una combinación de ambos. De esta manera, el carácter iónico parcial (el grado de polaridad), I, del enlace se podría calcular como 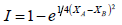 y la frontera entre los compuestos iónicos y los covalentes sería el 50% de ionicidad, con su correspondiente valor de 1.7 de diferencia de electronegatividades [27] -idea que, al no corresponder con la realidad, dejó de considerarse-. Más adelante, en 1937, para resolver el problema de la energía adicional proveniente de la resonancia iónica-covalente en los hidruros alcalinos, Pauling y Sherman [30] sugirieron usar el promedio geométrico en vez del aritmético, lo cual, en realidad, no se tomó en cuenta, tal vez porque, como dijo Derek Smith [31], no ayudaba mucho.
y la frontera entre los compuestos iónicos y los covalentes sería el 50% de ionicidad, con su correspondiente valor de 1.7 de diferencia de electronegatividades [27] -idea que, al no corresponder con la realidad, dejó de considerarse-. Más adelante, en 1937, para resolver el problema de la energía adicional proveniente de la resonancia iónica-covalente en los hidruros alcalinos, Pauling y Sherman [30] sugirieron usar el promedio geométrico en vez del aritmético, lo cual, en realidad, no se tomó en cuenta, tal vez porque, como dijo Derek Smith [31], no ayudaba mucho.
En 1939, Pauling propuso que "la electronegatividad es el poder de un átomo, en una molécula, para atraer electrones hacia él" [26]. Esta definición no estuvo exenta, al paso del tiempo, de algunas críticas. Por ejemplo, la de Sanderson (1986) indicando la derivación errónea de la electronegatividad de Pauling [32], comentario que provocó una respuesta aclaratoria defensiva, publicada en 1988 [33], o la de Allen (1990), calificando su primera propuesta de valores de electronegatividad como " un grupo de valores semicuantitativos para átomos libres, cuyas diferencias daban estimaciones numéricas aproximadas de polaridades " [34].
4.2. La cuantificación de la electronegatividad después de Pauling. 1ª mitad del siglo XX
Al plantear una escala de electronegatividad, el principal problema a resolver es predecir -o explicar- la distribución de la carga eléctrica en una molécula [23]. Al redefinir la electronegatividad de Pauling (XP), Mulliken, en 1934, propuso un método simple para cuantificarla [35] al usar el promedio aritmético entre los valores de dos propiedades atómicas: el primer potencial de ionización (I) y la afinidad electrónica (A). Así, la electronegatividad (o electroafinidad) de Mulliken (XM) puede definirse como la energía necesaria para atraer o soltar un electrón. Aunque el mismo Mulliken explicó el "empirismo" de su escala y lo "aproximado" de su definición, calificó a la primera de "absoluta", al no depender de una escala relativa arbitraria, como la de Pauling; sus unidades son las de la energía. Calculó los valores de electronegatividad para H, Li, B, C, N, O, F, Cl, Br, Be, e I, elementos de los que existían datos de afinidad electrónica. Estos valores serían recalculados por Parr y Pearson [36], en 1983, utilizando datos más precisos, aumentando el número de elementos a 25. A decir de Mullay, el método de Mulliken permite calcular los valores de la electronegatividad para los átomos en cualquier estado: neutro, excitado o con cualquier valencia, dándole la posibilidad de representar al átomo tal como existe en la molécula [23]. Sin embargo, Pearson advirtió, en 1990, que ni el potencial de ionización ni la afinidad electrónica correspondían a los átomos en estado neutro, sino a un estado de valencia "conveniente", lo que le permitió a Mulliken comparar su escala con la de Pauling, aunque el interés inicial de Mulliken estuviera en estudiar la polaridad del enlace [37]. Aunque se considera que la propuesta de Mulliken tiene una fundamentación teórica mayor que la de Pauling, su uso fue limitado debido a la falta de datos de afinidad electrónica -todavía en 2006 se conocían sólo los de 57 elementos-. Al contrastar su escala de electronegatividad (XM) con las de Malone (1933) [38], Pauling (1960) y Mulliken (1934), la encontró equivalente, aunque con valores 2.8 veces más grandes que la de Pauling: XP = 0.168 (XM - 1.23). Por lo que toca a las críticas, Allen señalaba "desórdenes severos obvios" en los valores de electronegatividad de Mulliken para algunos elementos: XH > XC; XH > XS; XCl > XO; y XBr > XO, lo que no se presenta en ninguna otra escala. Además menciona la "principal falla" de la escala: los valores de electronegatividad de los metales de transición se determinan solamente sobre la base de los electrones s más externos, sin considerar el número y comportamiento de los electrones d. Finalmente critica el sobredimensionamiento de la contribución aniónica que procede de considerar al mismo nivel A e I, cuando en la realidad tienen magnitudes diferentes [39].
En 1946 Walter Gordy propuso definir la electronegatividad de un átomo neutro, en una molécula estable, como la carga nuclear efectiva del átomo actuando sobre un electrón de valencia, a una distancia igual al radio covalente desde el núcleo: (Zeff)e/r, es decir, un potencial de superficie en los enlaces atómicos covalentes [40]. Basado en esta definición, Gordy también desarrolló una escala de electronegatividad. Boeyens [41] resalta que separar la capa de valencia del núcleo implica que la magnitud de la carga positiva efectiva del núcleo es igual a la carga electrónica de la capa de valencia. Gordy supuso un factor de apantallamiento constante, k, igual a 0.5 para cualquier electrón de la capa de valencia. Entonces Zeff = n - k(n - 1) = k(n + 1); donde n es el total de los electrones en la capa de valencia. Al sustituir 0.5(n + 1) por Zeff, en su propuesta, se obtiene que X = k(n + 1)/r, donde k = 0.5e. En el artículo referido, Gordy también señala que su modelo definía la electronegatividad absoluta de un elemento y que, en el caso de enlaces dobles o triples, la electronegatividad de un átomo dado sería mayor por tener un radio covalente menor. En la comparación de su escala con la de Pauling, encontró una gran diferencia para los valores de la plata, el oro y el cobre y lo explicó señalando que estos elementos contribuían con más de un electrón a los enlaces covalentes en sus compuestos, por lo que las constantes de apantallamiento debería tener un valor de 1 para todos los electrones, excepto para el electrón de la capa final. Cabe hacer notar que el valor de la carga efectiva en relación al radio (Zeff /r) es un potencial, mientras que la misma carga en relación al cuadrado del radio da como resultado una fuerza; seguramente esto fue notado y tomado en cuenta por Allred y Rochow, al realizar su trabajo [42]. Gordy comparó los valores de su escala (XG) con las de otros autores: Mulliken y Pauling: XP = 0.31XG + 0.50.
Sanderson es el autor de un método para la obtención de una escala de electronegatividad publicado en 1952. Se basó en el concepto de relación de estabilidad (SR, por sus siglas en inglés) [43]. SR es la relación de la densidad electrónica promedio (ED, por sus siglas en inglés) del átomo de un elemento activo entre la densidad electrónica de un átomo inerte (EDi). ED representa la compactación promedio de de la esfera electrónica respecto al núcleo atómico; se calcula como el número electrónico entre el volumen atómico, covalente o iónico: ED = Z/4.19r3. Por otro lado, SR es una medida de la compactación del átomo, de qué tan cerca y firmemente unidos están los electrones en el átomo. Sanderson ideó el número ED solo para propósitos de comparación, basándose en dos ideas que consideraba deberían ser tomadas en cuenta por toda explicación relativa a la electronegatividad: los átomos o iones que poseen electronegatividad tienen una atracción inherente por los electrones; y esos átomos o iones aumentan su estabilidad química después de aceptar los electrones. De allí que, si los átomos de todos los elementos tienen algo de electronegatividad, teóricamente poseen el poder de atraer a los electrones de otros átomos. Este poder es proporcional a la carga nuclear e inversamente proporcional al radio atómico y a la repulsión de la esfera electrónica del átomo. Entonces, la electronegatividad es aproximadamente proporcional a la densidad electrónica promedio, medida por medio de SR. Sanderson explicó el aumento en la estabilidad del átomo al ser anexados los electrones, definiendo el concepto de estabilidad máxima (SR = 1): al adquirir una carga negativa parcial la esfera electrónica se expande, lo cual se puede considerar "una mejora en el balance de fuerzas" al interior del átomo, disminuyendo la electronegatividad. Para este autor, el estudio de la naturaleza de los enlaces químicos y las propiedades de los compuestos eran las aplicaciones de la electronegatividad más evidentes. Más adelante (1986) propondría otra escala de electronegatividad [44] y, en 1988, la definiría "de manera menos controversial" como "la propiedad que determina la forma en que se distribuyen los electrones enlazantes entre uno y otro átomo" [45]. Al comparar la escala de Sanderson con la de Pauling se obtiene la ecuación XP = (0.2 (SR) + 0.77)2, una hipérbola tan abierta que parece una recta [23]. Allred y Rochow observaron que esta escala de electronegatividad mostraba una alternancia con el número atómico y que, en general, estaba de acuerdo con la de ellos (basada en la fuerza electrostática). También notaron que ese modelo podría explicar la alternancia en electronegatividad de grupos cercanos al centro de la Tabla Periódica, usando el empaquetamiento electrónico [42]. La escala de Sanderson no tuvo aceptación, posiblemente por su aparente oscuridad [23] o porque sus argumentos no fueron convincentes [46].
Sanderson también presentó una idea, a la que llamó igualación de la electronegatividad, para calcular su valor para un compuesto (desde una molécula diatómica hasta un sólido polimolecular) [47]. Básicamente establecía que los átomos del compuesto, inicialmente diferentes, terminan por tener la misma electronegatividad intermedia al constituirse el compuesto. Explicó que, para un compuesto binario, antes de formar un enlace, los átomos son eléctricamente neutros, pero mientras se forma el enlace, el átomo inicialmente más electronegativo gana más de la mitad de los electrones compartidos en él, quedando con una carga parcial negativa y menos electronegativo que al principio. El otro átomo, inicialmente menos electronegativo, queda con una carga parcial positiva, volviéndose más electronegativo. Estos cambios en la energía terminan cuando se alcanza el equilibrio y la electronegatividad se iguala en el compuesto. En este modelo, la electronegatividad es la media geométrica de las electronegatividades originales de los átomos [48]. Da como ejemplo, el cálculo de la electronegatividad del compuesto CH3COCl -usando las XM-:
[(XC)2 * (XH)3 * (XO) * (XCl)] 1/7 = (2.746 * 2.5923 * 3.654 * 3.475)1/7 = 2.886.
A pesar de los argumentos planteados por Sanderson, este formalismo tampoco tuvo mucha aceptación, hasta que Robert Parr y coautores publicaron un artículo cuya hipótesis era la equivalencia entre la electronegatividad y el negativo del potencial químico electrónico (-m) [49]; sin embargo, cuando se encontró que eran similares, pero no iguales [34], la idea perdió nuevamente popularidad. Sumado a esto, Allen enumeró cinco "severas" objeciones a la hipótesis de la igualación de la electronegatividad -y a otros postulados establecidos por Sanderson- [41], que van más allá de los objetivos de este trabajo. No obstante todo lo anterior, algunos autores siguen aceptando el formalismo de Sanderson, como Noorizadeh y Shakerzadeh [50], últimos en proponer una escala de electronegatividad basada en los valores calculados de electrofilicidad.
La escala de Albert L. Allred (1931-?) y Eugene G. Rochow (1909-2002) [42], tiene la misma base teórica que la de Gordy [40]. Para estos autores la electronegatividad debería relacionarse a la carga experimentada por un electrón en la superficie de un átomo, de manera que cuanto mayor fuera la carga por unidad de superficie atómica, mayor sería la tendencia del átomo para atraer electrones, es decir, una fuerza electrostática calculada como F = e2Zeff /r2, donde r es el radio covalente y eZeffla carga nuclear efectiva. La plantearon como una aproximación a la fuerza con la que un átomo, en una molécula, atrae electrones y la llamaron electronegatividad absoluta del átomo. Calcularon la carga nuclear efectiva usando las reglas de Slater y supusieron que el área superficial del átomo en una molécula podía ser tomada como proporcional al cuadrado del radio covalente, si éste se expresaba en Angstroms. De esta manera, la electronegatividad dependía mayormente de la combinación de los orbitales usados para el enlace y, como la mayoría de los elementos exhibían más de un tipo de hibridación en sus compuestos, sería más adecuado hablar de una electronegatividad orbital y asignarle un intervalo de valores a un elemento y no tratarla como una propiedad atómica invariante. Allred y Rochow consideraron sus propuestas totalmente consistentes con la definición de Pauling, pues estimaba, precisamente, la tendencia de los átomos a atraer electrones. Existe una particularidad en esta escala, ya observada por Sanderson [43]: la electronegatividad del Ge es mayor que la del Si, lo que difiere de la de Pauling [51]. Compararon su escala con la de Gordy [40], la de Pritchard [46] y, la de Pauling XP = 0.359 XA-R + 0.744.
En la 2ª mitad del siglo XX vendrían otras propuestas.
5. Conclusión
Ninguno de los caminos seguidos por los químicos del siglo XIX para entender la afinidad fructificó en una idea clara ni llevó a una posible cuantificación. Fue sólo hasta el siglo XX, con Pauling, que esto comenzó a suceder, aunque el nombre del concepto afinidad química entró en desuso; sin embargo, la esencia de lo que representaba continuó vigente. El descubrimiento y desarrollo de la electricidad, la termodinámica, y la estructura del átomo fueron fundamentales para que el concepto de electronegatividad (la afinidad) encontrara nuevas vetas de perfeccionamiento de lo que es y de su medición, lo cual promovió una mayor discusión y, por ende, la reflexión y el planteamiento de nuevas propuestas.
Referencias
[1] White, M., Isaac Newton: The Last Sorcerer, Londres, Fourth Estate, 3 P, 1977. [ Links ]
[2] Hampson, N., The Enlightenment, Londres, Penguin, 458 P, 2003. [ Links ]
[3] Gleick, J., Isaac Newton, Londres, Fourth Estate, pp. 101-108, 2003. [ Links ]
[4] Frank, E. Manuel, A Portrait of Isaac Newton, Cambridge, Harvard University Press, 398 P, 1968. [ Links ]
[5] Salas, G., Ramírez, J., Restrepo, O., Noguez, M y Cockrell, B., La Importancia de llamarse afinidad química. Parte IV: Las primeras flores. DYNA, 81 (184), pp. 225-232, 2014. [ Links ]
[6] Real Academia de Ciencias Exactas, Físicas y Naturales, Diccionario esencial de las ciencias. España, Espasa, p. 29, 2001. [ Links ]
[7] Salas, G., Ramírez, J., Restrepo, O., Cockrell, B. y Noguez, M., La Importancia de llamarse afinidad química. Parte I: La semilla. DYNA, 79 (173), pp. 135-144, 2012. [ Links ]
[8] Salas, G., Ramírez, J., Restrepo, O., Noguez, M. y Cockrell, B., La Importancia de llamarse afinidad química. Parte II: La semilla germina. DYNA, 80 (177), pp. 162-170, 2013. [ Links ]
[9] Salas, G., Ramírez, J., Restrepo, O., Noguez, M. y Cockrell, B., La Importancia de llamarse afinidad química. Parte III: El crecimiento vano. DYNA, 80 (181), pp. 219-227, 2013. [ Links ]
[10] Berthollet, C.L., Essai de Statique Chimie, Paris, Demonville et Soeurs, 1803. [ Links ]
[11] Davy, H., On some Chemical Agencies of Electricity, Phil. Tras. R. Soc. Lond., 97, pp. 1-56, 1807. [ Links ]
[12] Avogadro, A., Idées sur l'acidité et l'alcalinité, Journal de physique, de chimie, d'histoire naturelle et des arts, Tome LXIX, pp. 142-148, 1809. [ Links ]
[13] Berzelius, J.J., Lehrbuch der Chemie, Dresde, Arnoldi, 1825. [ Links ]
[14] Jensen, W.B., Electronegativity from Avogadro to Pauling. Part I: Origins of the Electronegativity Concept, Journal of Chemical Education, 73 (1), pp. 11-20, 1996. [ Links ]
[15] Dalton, J., A new system of chemical philosophy, Londres , S. Russell, 1808. [ Links ]
[16] Barker, G.F., A Text-book of elementary chemistry, Louisville, John P. Morton & Co, 1870. [ Links ]
[17] Cook, J.P., The New Chemistry, New York, D. Appleton & Co., 1876. [ Links ]
[18] Van't Hoff, J. and Thomson, J., Lectures on theoretical and physical chemistry, Londres, Edward Arnold, 1899. [ Links ]
[19] Caven, R.M. and Lander, G.D., Systematic inorganic chemistry from the standpoint of the periodic law, Londres, Blackie & son, 1906. [ Links ]
[20] Sackur, O., A Text book of Thermo-Chemisty and Thermodynamics, Londres, Macmillan & Co, 1913. [ Links ]
[21] Von Helmholtz, H., On the modern development of Faraday's conception of electricity, J. Chem. Soc. Trans., 39, pp. 277-304, 1881. [ Links ]
[22] Jensen, W.B., Electronegativity from Avogadro to Pauling. Part II: Late nineteenth- and early Twentieth-Century developments, Journal of Chemical Education, 80 (3), pp. 279-287, 2003. [ Links ]
[23] Mullay, J., Estimation of atomic and group electronegativities, In Sen, K. and Jørgensen, C.K. (eds), Electronegativity, Heidelberg, Springer-Verlag, 1987, pp. 1-26. [ Links ]
[24] Pauling, L. and Yost, D.M., The additivity of the energies of normal covalent bonds, Proceedings of the National Academy of Sciences, 18, pp. 414-416, 1932. [ Links ]
[25] Bergmann, D. and Hinze, J., Electronegativity and molecular properties, Angewandte Chemie Edición Internacional, 35, pp. 150-163, 1996. [ Links ]
[26] Pauling, L., The nature of chemical bond and the structure of molecules and crystals: An introduction to modern structural chemistry, Ithaca, Cornell University Press, 1939. [ Links ]
[27] Pauling, L., The nature of chemical bond, Ithaca, Cornell University Press, 1960. [ Links ]
[28] Allred, A.L., Electronegativity values from thermochemical data, Journal of Inorganic & Nuclear Chemistry, 17, pp. 215-221, 1961. [ Links ]
[29] Fung, B., The electronegativity of noble gases, Journal of Physical Chemistry, 69 (2), pp. 596-600, 1965. [ Links ]
[30] Pauling, L. and Sherman, J., A quantitative discussion of bond orbitals, Journal of the American Chemical Society, 59 (8), pp. 1450-1456, 1937. [ Links ]
[31] Smith, D.W., Comment on "Evaluation and test of Pauling's electronegativity scale", Journal of Physical Chemistry A, 106, pp. 5951-5952, 2002. [ Links ]
[32] Sanderson, R.T., Is the theoretical emperor really wearing any clothes?, Journal of Chemical Education, 63 (10), pp. 845-846, 1986. [ Links ]
[33] Pauling, L., The origin and nature of the electronegativity scale, Journal of Chemical Education, 65 (4), p. 375, 1988. [ Links ]
[34] Allen L.C., Electronegativity scales, Accounts of Chemical Research, 23, pp. 175-176, 1990. [ Links ]
[35] Mulliken, R.S., A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities, Journal of Chemical Physics, 2, pp. 782-793, 1934. [ Links ]
[36] Parr, R.G. and Pearson, R.G., Absolute Hardness: Companion parameter to absolute electronegativity, Journal of the American Chemical Society, 105, pp. 7512-7516, 1983. [ Links ]
[37] Pearson, R.G., Electronegativity scales, Accounts of Chemical Research, 23 (1), pp. 1-2, 1990. [ Links ]
[38] Malone, J.G., The electric moment as a measure of the ionic nature of covalent bonds, Journal of Chemical Physics, 1, pp. 197-199, 1933. [ Links ]
[39] Allen, L.C., Chemistry and electronegativity, International Journal of Quantum Chemistry, 49, pp. 253-277, 1994. [ Links ]
[40] Gordy, W., A relation between bond force constants, bond orders, bond lengths, and the electronegativities of bonded atoms, Journal of Chemical Physics, 14 (5), pp. 305-320, 1946. [ Links ]
[41] Boeyens, J.C.A., The periodic electronegativity table, Zeitschrift für Naturforschung, 63b, pp. 199-209, 2008. [ Links ]
[42] Allred, A.L. and Rochow, E.G., A scale of electronegativity based on electrostatic force, Journal of Inorganic & Nuclear Chemistry, 5, pp. 264-268, 1958. [ Links ]
[43] Sanderson, R.T., An explanation of chemical variations within periodic major groups, Journal of the American Chemical Society, 74 (19), pp. 4792-4794, 1952. [ Links ]
[44] Sanderson, R.T., Electronegativity and bonding of transitional elements, Inorganic Chemistry, 25, pp. 3518-3522, 1986 [ Links ]
[45] Sanderson, R.T., Principles of electronegativity. Part I. General nature, Journal of Chemical Education, 65 (2), pp. 112-118, 1988. [ Links ]
[46] Pritchard, H.O. and Skinner, H.A., The concept of electronegativity, Chemical Reviews, 55, pp. 745-786, 1955. [ Links ]
[47] Sanderson, R.T., An interpretation of bond lengths and a classification of bonds, Science, New series, 114 (2973), pp. 670-672, 1951. [ Links ]
[48] Sanderson, R.T., Principles of electronegativity. Part II. Applications, Journal of Chemical Education, 65 (3), pp. 227-231, 1988. [ Links ]
[49] Parr, R.G., Robert A., Donnelly, R.A., Levy, M. and Palke, W.E., Electronegativity: The density functional viewpoint, Journal of Chemical Physics, 68 (8), pp. 3801-3807, 1978. [ Links ]
[50] Noorizadeh, S. and Shakerzadeh, E., A new scale of electronegativity based on electrophilicity index, Journal of Physical Chemistry, 112, pp. 3486-3491, 2008. [ Links ]
[51] Pauling, L., Atomic radii and interatomic distances in metals, Journal of the American Chemical Society, 69 (3), pp. 542-553, 1947. [ Links ]
G. Salas-Banuet, Ing. Químico Metalúrgico con título de Postgrado, ambos de la Universidad Nacional Autónoma de México (UNAM), México. Profesor Titular "C" en la Facultad de Química de la misma universidad, donde ha trabajado en docencia, investigación y divulgación por 39 años, de tiempo completo. Su principal área de interés es la enseñanza. Su área principal de trabajo está en los materiales, especialmente metales, las relaciones estructura-propiedades, nuevos procesos de obtención, diseño y reemplazamiento de aleaciones, reciclaje, el desarrollo y el impacto de la ciencia y la tecnología; otros campos de interés son el pensamiento, las ideas, la historia, sociología, arqueología, desarrollo humano, leyes, economía, funcionamiento cerebral, entre otros. Ha enseñado en cursos de pregrado y postgrado (más de 165), publicado en revistas nacionales (más de 30) e internacionales indexadas (más de 20). Así mismo ha enviado trabajos a conferencias y eventos científicos internacionales (más de 80) y nacionales (más de 25) y ha sido conferencista invitado en más de 25 oportunidades, algunas de ellas de carácter magistral.
J. Ramírez-Vieyra obtuvo su diploma en Ingeniería Metalúrgica en 1992 en la Universidad Nacional Autónoma de México (UNAM), México, donde también obtuvo su MSc en Ingeniería. Fue profesor adjunto (1993-1994) y ha sido Investigador Asistente. Adicionalmente, actúa como Profesor Asistente en la Facultad de Química de la UNAM. Ha colaborado en algunos proyectos de investigación en la elaboración de artículos y ha participado en alrededor de 50 congresos y ha publicado cerca de 20 artículos en sus temas de interés: Metalurgia física, Arqueometalurgia, Educación en ingeniería y Materiales y ambiente.
O. Restrepo-Baena, recibió su título de Ing. de Minas y Metalurgia en 1991 en la Universidad Nacional de Colombia. MSc. en Evaluación de Impactos Ambientales del Instituto de Investigaciones Ecológicas de Málaga, España y Dr. en Metalurgia y Materiales de la Universidad de Oviedo, España. Trabaja en la Facultad de Minas de la Universidad Nacional de Colombia, Sede Medellín desde 1997 y su área de trabajo se centra en Metalurgia extractiva y Materiales de Ingeniería, Cementos, Cerámicos, Pigmentos.
M. Noguez-Amaya, es Ing. Química Metalúrgica de la Facultad de Química, Universidad Nacional Autónoma de México (UNAM), México, 1964-1968. MSc. en Ingeniería Metalúrgica de la School of Engineering, University of Pittsburgh, Pittsburgh, Pa., USA, 1973. Ha trabajado profesionalmente en la industria automotriz y en la industria de fundición como Ingeniera de Manufactura 1968-1970. En la UNAM, ha sido profesora desde 1968 Profesora Asociada en 1974 y Profesora en 1984. Actualmente profesora de tiempo completo en la Facultad de Química de la UNAM, donde ha sido miembro del Consejo Técnico primero como estudiante y después como profesora por 3 periodos (1976-2001); coordinadora de los estudios de pregrado en Ingeniera Química Metalúrgica (1979-1981); ganó el premio "Ernesto Rios del Castillo" (1995-1997), y el premio "Sor Juana Inés de la Cruz" a la mujer universitaria de la UNAM (2013), de la Escuela de Química. Sus áreas de interés son: Metalurgia física, Educación en ingeniería y Materiales y ambiente.
B. R. Cockrell, recibió su PhD. en Antropología en 2014 (mención en Arqueología) en la University of California, Berkeley, USA Su área de investigación actual está en el análisis de metales del Cenote Sagrado de Chichen Itza en Yucatán, México, en colaboración con investigadores de la UNAM. Actualmente enseña en el Departamento de Antropología de UC Berkeley y San Francisco State University, USA. Sus intereses incluyen la metalurgia de los antiguos pueblos americanos así como la arqueometría y arqueología de Mesoamérica.
