










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkDYNA
versão impressa ISSN 0012-7353
Dyna rev.fac.nac.minas vol.82 no.189 Medellín jan./fev. 2015
https://doi.org/10.15446/dyna.v82n189.41963
http://dx.doi.org/10.15446/dyna.v82n189.41963
Insights of asphaltene aggregation mechanism from molecular dynamics simulation
Visualización del mecanismo de agregación de asfaltenos usando simulación de dinámica molecular
Jennifer De León a, Bibian Hoyos b & Wilson Cañas-Marín c
a Facultad de Minas, Universidad Nacional de Colombia, Medellín, Colombia. jde@unal.edu.co
b Facultad de Minas, Universidad Nacional de Colombia, Medellín, Colombia. bahoyos@unal.edu.co
c ECOPETROL S.A., Instituto Colombiano del Petróleo (ICP), km 7 via Piedecuesta, Santander, Colombia, wilson.cmarin@ecopetrol.com.co
Received: February 7th, 2014. Received in revised form: September 10th, 2014. Accepted: October 1th, 2014.
Abstract
Asphaltene aggregation process was studied using molecular dynamics techniques. Four different structures were used. The first three molecules have a continental structure, with condensed aromatic cores, while the forth has an archipelago structure, with small groups of aromatic rings connected with saturated chains. The molecules were constructed in an atomistic framework, in which atoms are described individually. Interaction forces were calculated at 300 K and 200 atm; Van der Waals and electrostatic interactions were evaluated separately. For all four molecules the solubility parameter was calculated. It was found that Van der Waals interactions due to the presence of aromatic rings and electrostatic forces caused by the presence of heteroatoms such as oxygen, nitrogen and sulfur, are equally relevant in the aggregation of asphaltene molecules. For all molecules it was found asphaltene systems are more stable in aggregation state than in monomeric state. For continental structures, the presence of long ramifications obstructs the formation of asphaltene aggregates. For archipelago structures, the flexibility of the molecules enables the aggregation with other structures. The presence of heteroatoms creates a repulsive force that hinders the aggregation process. The molecular volume and the cohesive energy are also sensitive to the geometric configuration and the composition of the species, which affects the solubility parameter.
Keywords: asphaltene, aggregation, molecular simulation, solubility parameter.
Resumen
Se estudió el proceso de agregación de asfaltenos utilizando técnicas de dinámica molecular. Se utilizaron cuatro estructuras diferentes. Las primeras tres moléculas tienen una estructura continental, con núcleos aromáticos condensador, mientras que la cuarta posee una estructura tipo archipiélago, con pequeños grupos de anillos aromáticos conectados con cadenas saturadas. Las moléculas fueron construidas de manera atomística, en la cual cada átomo se describe individualmente. Se calcularon las fuerzas de interacción a 300 K y 200 atm; las fuerzas de Van der Waals y las interacciones electrostáticas fueron evaluadas separadamente. Se calculó el parámetro de solubilidad para las cuatro moléculas. Se encontró que las interacciones de Van der Waals asociadas a los anillos aromáticos y las fuerzas electrostáticas ocasionadas principalmente por la presencia de heteroátomos como oxígeno, azufre y nitrógeno, son igualmente relevantes en la agregación de moléculas de asfalteno. Para todas las moléculas se encontró que los sistemas de asfaltenos tienen menor energía en estado de agregación que en estado monomérico. Para las estructuras continentales, la presencia de largas cadenas obstruye el proceso de formación de agregados. Para las estructuras tipo archipiélago, la flexibilidad de las moléculas facilita la agregación con otras estructuras. La presencia de heteroátomos ocasiona una fuerza repulsiva que dificulta la agregación. El volumen molecular y la energía de cohesión también son sensibles a la configuración geométrica y la composición de las especies, lo cual afecta el parámetro de solubilidad.
Palabras clave: asfaltenos, agregación, simulación molecular, parámetro de solubilidad.
1. Introduction
Asphaltene aggregation and deposition is one of the biggest sources of production loss, especially in light oil reservoirs. However, the real mechanism of asphaltene aggregation is still unclear. Due to the complexity of the system, it is particularly difficult to design proper experiments in order to determine the exact nature of the aggregation process.
Molecular simulation techniques appear as an alternative to experimentation, as these allow, for instance, studying the behavior of complex systems under extreme conditions of temperature and pressure. In the literature, it is possible to differentiate three major areas in computer simulation of asphaltene systems: (1) the development of adequate equations of state to describe hydrocarbon systems with some contents of asphaltene [1-3]; (2) population balance models that study the evolution of aggregate size and growth as a function of some control variable; and (3) molecular models to study the behavior of asphaltene systems from a molecular or atomistic scale [4-8].
With the improvements in computer efficiency, molecular models become an excellent choice for accurately describing the aggregation of asphaltenes, as this process takes place in a molecular scale. These models are often based on experimental measurements and theoretical considerations, taking into account the average composition as a restriction.
Vicente et al. [4] presented a model to calculate the miscibility of a model asphaltene molecule in different organic solvents, finding that asphaltenes have more affinity with long-chain hydrocarbons due to the density of this kind of substances, which agrees with experimental results. Lu et al. [5] evaluated the aggregation of asphaltene molecules in different solvents at various temperatures. Results showed that temperature does not influence aggregation up to 500 K. Frigeiro and Molinari [6] used molecular dynamics to study aggregation in different solvents and calculated the solubility parameter of two asphaltene molecules proposed after experimentation and spontaneous formation of asphaltene clusters was observed. A. N. M. Carauta et al. [7] also used molecular dynamics to calculate the solubility parameter of an asphaltene molecule and its dimer. Their results show that the solubility parameter decreases with aggregation number. More recently, M. Segdhi et al. [8] studied the aggregation of asphaltenes and the formation of dimers and clusters; their results show the different states of aggregation and were able to calculate aggregate sizes. All these results allow concluding that computer simulation is an adequate technique to describe asphaltene systems and their behavior.
Although it might seem that computer simulation of asphaltene-petroleum systems holds all the answers in understanding asphaltene behavior and prediction of macroscopic properties, one important aspect to consider in using this technique is that they need the molecular description of asphaltene as input, and several aspects of asphaltene structure and composition are still in debate.
Asphaltenes are not a single specific molecule, but rather a set of structures that share solubility behavior and general characteristics. Traditionally, structural characteristics are determined experimentally, and average values for atomic content, molecular weight and geometric configuration are used to propose one structure to represent the different molecules that coexist in the actual asphaltene system [9,10].
This representation of asphaltenes as a single average molecule has recently originated some controversy. Gray et al. [11] have stated that as different structures exist in the actual system, the asphaltene model should include all these structures to adequately describe asphaltene aggregation behavior.
In light of this new tendency, four asphaltene molecules obtained from previous experimental work in Colombian Castilla crude oil were studied. Since all four molecules were obtained from experimental work, it is possible that they all coexist in the actual asphaltene-hydrocarbon system. The aim of this work is to determine the nature of the behavior of these molecules as they interact with themselves in order to set a foundation in asphaltene aggregation studies of Colombian asphaltene systems. The molecular volume and the solubility parameter were also calculated to validate the model with macroscopic properties.
2. Model
The asphaltene molecules used in this study were proposed by Navarro et al. [12] as a result of experimental analyses conducted on Colombian Castilla crude oil. In the original work presented by Navarro et al. three molecules were proposed, one asphaltene macromolecule and two molecules presented as resins. However, the recent literature on asphaltenes structure and new measurements of properties such as molecular mass and density for asphaltene monomeric molecules suggest that the macromolecule initially proposed by Navarro et al. as an asphaltene does not meet the criteria of an asphaltene molecule [9,10,13], and the initially proposed resins can be categorized as asphaltenes. In this work, a decision was made to divide the macromolecule proposed by Navarro et al. into two molecules, and to treat the proposed resins as asphaltene molecules. The four proposed asphaltene molecules are likely to coexist in the actual hydrocarbon system. However, in this work they are evaluated individually in order to establish the true nature of their aggregation behavior in absence of external forces.
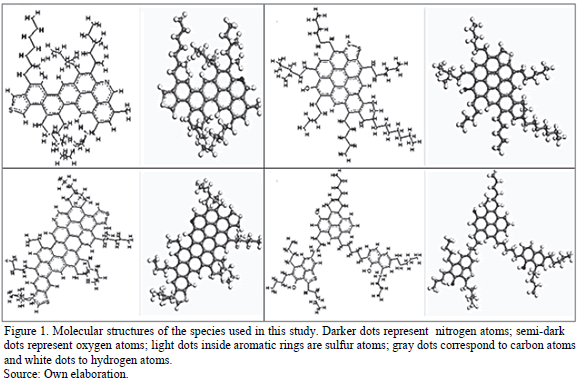
Fig. 1 shows the molecular structure of the species used in this study. It is seen that the structures shown in Fig. 1a, 1b and 1c have a continental structure, with a core made of condensed aromatic rings surrounded by saturated chains of hydrocarbons. The fourth molecule, shown in Fig. 1d, has an archipelago structure, with small groups of rings connected by saturated chains. Molecule A1 (Fig. 1a) presents a core made of eight aromatic rings and one atom of sulfur and one atom of nitrogen. Molecule A2 (Fig. 1b) has a similar structure, but has a larger core made of 15 aromatic rings, and one more atom of sulfur. Molecule A3 (Fig. 1c) has a condensed core consisting on nine aromatic rings, but has an oxygen and a sulfur atom, and no nitrogen. This molecule also has longer saturated ramifications on its structure. Molecule A4 (Fig. 1d) has three aromatic groups connected with saturated chains and it also has three oxygen atoms and one sulfur atom, and no nitrogen.
The molecules were represented in an atomistic framework, in which each atom is considered individually using the LAMMPS package and the consistent valence force field CVFF [14]. The interaction energy was evaluated as stated in eq. (1).
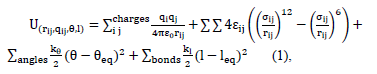
Where e and s are the Lennard-Jones potential parameters, 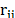 is the separation distance between atoms,
is the separation distance between atoms, 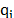 and
and 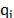 are the atomic charges;
are the atomic charges; 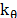 ,
, 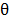 and
and 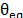 are the angle constant, the angle between each three atoms and the equilibrium angle, respectively; and
are the angle constant, the angle between each three atoms and the equilibrium angle, respectively; and 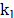 ,
, 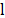 and
and 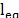 are the bond constant, the bond length between each pair of atoms and the equilibrium bond length, respectively.
are the bond constant, the bond length between each pair of atoms and the equilibrium bond length, respectively.
Lennard-Jones parameters and bond and angle constants were assigned considering the nature of the atoms that form the pair, bond or angle, respectively. Partial charge for each atom was assigned using Equivalent Charge method [15].
In order to determine the manner in which asphaltenes form aggregates, the interaction energy is separated into its different contributions; only non-bond energy is considered, that is, Van der Waals and electrostatic energy.
The solubility parameter was calculated using the Scatchard-Hildebrand formulation, and it is stated in eq. (2).

where 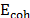 is the cohesive energy of each molecule, and
is the cohesive energy of each molecule, and 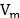 is its molar volume. The cohesive energy is the energetic difference between the ideal gas state and the condensed state. To calculate the cohesive energy, the molecules were simulated under two conditions. First, the species were isolated to emulate the ideal gas state. Second, the molecules were surrounded by replicas of themselves in order to depict the condensed state. Finally, the molecular volume is also calculated using molecular simulation.
is its molar volume. The cohesive energy is the energetic difference between the ideal gas state and the condensed state. To calculate the cohesive energy, the molecules were simulated under two conditions. First, the species were isolated to emulate the ideal gas state. Second, the molecules were surrounded by replicas of themselves in order to depict the condensed state. Finally, the molecular volume is also calculated using molecular simulation.
3. Technical details
In order to replicate the reservoir conditions, and at the same time compare results with previous work [6,7], a temperature of 300 K and a pressure of 200 atm were selected. For each species, a NPT simulation box was built, so as to maintain the number of particles, the pressure and the temperature of the system.
The cut radius for each species was selected as the length of the molecule for guaranteeing adequate distance of interaction between the molecules. This length was calculated as the distance, in Angstroms, between the further pair of atoms. The Ewald's summation method was used to evaluate the long range electrostatic interactions. The SHAKE algorithm was used to maintain bond length. A complete list of the parameters used in this simulation can be found in [16].
For each structure in Fig. 1, an energy minimization was conducted in order to find the proper geometric configuration in which the molecule had more stability. This minimization consists on taking the molecule to a temperature of 0 K and allowing it to relax and finding the most adequate configuration, using a modification of the conjugate gradient algorithm available in the LAMMPS package [14]. The tolerance for this process was set to 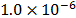 kcal/mol. Once the minimum energy state was achieved; the molecules were taken to the work temperature and pressure. Once this state was reached, two simulations were conducted; the first one consisted on simulating ideal gas state, for which each molecule was placed in a simulation box with reflective walls to avoid interactions outside the molecule itself in order to calculate the cohesive energy of the isolated monomer. The second simulation consisted on simulating the condensed state, for which the molecule is surrounded by twelve replicas of itself and placed in a simulation box with periodic boundary conditions in all three dimensions. The number of replicas was chosen arbitrarily, considering both the accuracy of the measurement and maintaining reasonable computing time. This simulation allows calculating the molecular volume of the monomer and the cohesive energy of the molecules in condensed state at working pressure and temperature.
kcal/mol. Once the minimum energy state was achieved; the molecules were taken to the work temperature and pressure. Once this state was reached, two simulations were conducted; the first one consisted on simulating ideal gas state, for which each molecule was placed in a simulation box with reflective walls to avoid interactions outside the molecule itself in order to calculate the cohesive energy of the isolated monomer. The second simulation consisted on simulating the condensed state, for which the molecule is surrounded by twelve replicas of itself and placed in a simulation box with periodic boundary conditions in all three dimensions. The number of replicas was chosen arbitrarily, considering both the accuracy of the measurement and maintaining reasonable computing time. This simulation allows calculating the molecular volume of the monomer and the cohesive energy of the molecules in condensed state at working pressure and temperature.
Molecular dynamic simulations of at least 10 ns were conducted, using a timestep of 1 fs. Non-bond energies (Van der Waals and electrostatic) were evaluated. Molecular volume was estimated and the solubility parameter was calculated for all four species.
4. Results and discussion
Fig. 2 shows the results for non-bond energy of all molecules. Fig. 2.a depicts energy results for molecule A1 in isolated conditions and Van der Waals, electrostatic and total non-bond energy for periodic conditions after an equilibration period of 9.5 ns. It is also evident from this figure, the molecule has lower energy when is surrounded by its replicas than in isolated conditions (i.e., as a monomer). This is quite obvious, considering the isolated case resembles ideal gas conditions. However, this results show that the model here proposed is able to replicate the aggregation behavior observed experimentally in asphaltenes molecules [6]. It is worth noting that, even though asphaltenes do not exhibit polymeric behavior, the term "monomer" has gained acceptance in the literature to refer to asphaltenes in a disaggregated state.
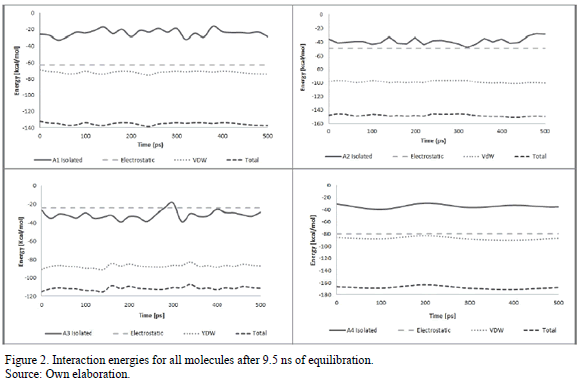
As to the aggregation mechanism, it can also be seen from Fig. 2.a that the Van der Waals energy, associated with the aromatic content of the molecule, and the electrostatic energy, associated with the presence of heteroatoms, are comparable in magnitude, even though for molecule A1 the Van der Waals energy is higher in magnitude. That is, the effect of the continental structure and the aromatic core is equally important as the electrostatic effect for presence of heteroatoms, so neither one can be neglected.
Fig. 2.b shows the energetic results for species A2 after 9.5 ns of equilibration. Again, it can be seen that the molecule has lower energy in the aggregate state, that is, is more stable. In this case, the Van der Waals energy has a value that is higher in magnitude than the electrostatic energy. This can be caused by the fact that the A2 molecule has more aromatic rings in its structure and fewer heteroatoms.
Fig. 2.c shows the energetic results for molecule A3 after 9.5 ns of equilibration. Similar to the previous cases, the total energy of the condensed state is lower than the isolated state, consistent with its tendency to form aggregates. However, for this molecule the total energy in the aggregated state (the sum of Van der Waals and electrostatic contributions) is higher than for the other continental structures. While molecule A3 still has a continental structure, it also has longer saturated chains, which appear to create a physical barrier that challenges the aggregation process. For this particular case, the difference between electrostatic interactions and Van der Waals interactions is more pronounced. This is associated with the oxygen atom attached to the continental structure. While the aromatic rings contribute to the aggregation process, the presence of oxygen, which has a greater partial charge than nitrogen or sulfur, seems to make it more difficult for the rigid molecule to balance the charges. However, like in the previous cases, the electrostatic interaction still has a negative energy value and therefore contributes to the aggregation process, and should not be neglected.
Fig. 2.d presents the energetic results for molecule A4. As for all other molecules, these results corroborate the aggregation behavior. For this monomeric molecule, the Van der Waals and the electrostatic contributions are similar, probably due to the fact that the presence of oxygen, with a high partial charge, and the dispersed aromatic cores contribute in equal manner to the formation of aggregates. Molecule A4 exhibits lower energy in aggregate conditions than the other molecules, which can be explained because this molecule has a different, more flexible, structure that allows for better interaction with other molecules by steric ability.
The behavior of the difference between Van der Waals and electrostatic energies was similar for pairs A1-A3 and A2-A4. This was somewhat surprising considering molecule A4 has an archipelago structure that differs from the other three continental ones. This is evidence that not only molecular architecture (island or archipelago) or heteroatoms content are crucial when defining asphaltene interactions, but the aliphatic chains also play a definitive role in aggregation.
As an example, Fig. 3 shows the final configuration for the interaction of molecule A2 with itself. As can be seen from the graphic, the aromatic cores locate in parallel while the side chains locate in the periphery.
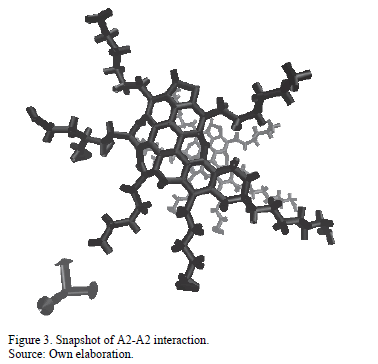
This is consistent with the behavior observed in the energy results. Van der Waals interactions, associated with the aromatic core, and electrostatic interactions, associated with the presence of heteroatoms mostly, contribute in similar way to the aggregation process, as the molecule experiences a strong attraction in the aromatic core while balances the charges attached to the heteroatoms.
Fig. 4 contains a snapshot of the final configuration of the interaction of molecule A3 with itself. This figure illustrates the behavior of long chains of hydrocarbons in the asphaltene structure as they suppose an obstacle to the formation of aggregates. This behavior is consistent with the energy results obtained for molecule A3, as this molecule has higher energy in aggregated state compared with the other continental structures.
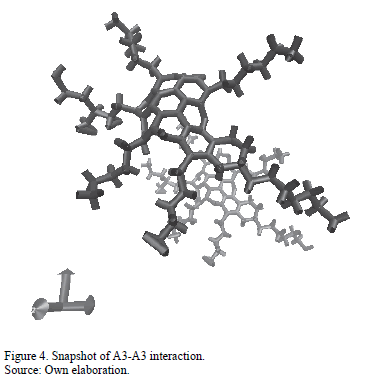
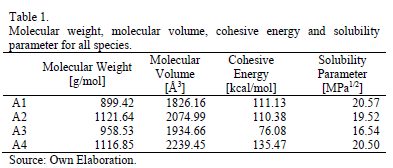
Table 1 summarizes the results obtained for molecular volume, cohesive energy and solubility parameter of all four molecules. The reported experimental values for asphaltene solubility parameter range between 17.0 and 21.0 MPa1/2 [6,7,13,17]. It can be seen that the asphaltenes here presented are within expected values for this parameter. For molecule A3, the solubility parameter is slightly lower than expected; this is due to the fact that the repulsive forces derived from the long saturated chains make the energy difference between isolated and periodic state to be lower in magnitude than for the other molecules.
5. Conclusions
Asphaltene aggregation is a complex process, influenced by many factors. Van der Waals interactions due to the presence of aromatic rings and electrostatic forces caused by the presence of heteroatoms such as oxygen, nitrogen and sulfur, are equally relevant in the aggregation of asphaltene molecules. For the molecules here studied, the model successfully reproduced the expected aggregation behavior, as the lower energy in this state so indicated. The structure and elemental composition of asphaltene are also crucial in determining the manner in which asphaltenes aggregate. The presence and geometrical distribution of heteroatoms with distinct partial charges influences the formation and stability of the aggregates. For continental structures, the presence of long ramifications obstructs the formation of asphaltene aggregates. For archipelago structures, the flexibility of the molecules enables the aggregation with other structures. The presence of heteroatoms creates a repulsive force that hinders the aggregation process. The molecular volume and the cohesive energy are also sensitive to the geometric configuration and the composition of the species, which affects the solubility parameter. The solubility parameter was within the accepted range for all molecules. Finally, the molecular simulation techniques seem to be adequate to describe asphaltene structure and aggregation behavior. This work is preliminary in understanding Colombian asphaltene behavior. Further work must be conducted in search of a more general understanding, simulating asphaltene aggregation in various solvents and linking results with experimental findings.
References
[1] Punnapala, S. and Vargas, F.M., Revisiting the PC-SAFT characterization procedure for an improved asphaltene precipitation prediction. Fuel, 108, pp. 417-429, 2013. http://dx.doi.org/10.1016/j.fuel.2012.12.058. [ Links ]
[2] Wu, J., Prausnitz, J.M. and Firoozabadi, A., Molecular-thermodynamic framework for asphaltene-oil equilibria. AIChE Journal, 44 (5), pp. 1188-1199, 1998. [ Links ]
[3] Artola, P.A., Pereira, F.E., Adjiman, C.S., Galindo, A., Müller, E.A., Jackson, G. and Haslam, A.J., Understanding the fluid phase behaviour of crude oil: Asphaltene precipitation. Fluid Phase Equilbiria. 306 (1), pp. 129-136, 2011. http://dx.doi.org/10.1016/j.fluid.2011.01.024. [ Links ]
[4] Vicente, L., Soto, C., Pacheco-Sánchez, H., Hernández-Trujillo, J. and Martinez-Magadán, J.M., Application of molecular simulation to calculate miscibility of a model asphaltene molecule. Fluid Phase Equilibria. 239 (1), pp. 100-106, 2006. http://dx.doi.org/10.1016/j.fluid.2005.11.001 [ Links ]
[5] Lu, G., Li, Y., Song, H., Yu, Y. and Wang. C., Micromechanism of petroleum asphaltene aggregation. Petroleum Exploration and Development. 35 (1), pp. 67-72, 2008. [ Links ]
[6] Frigerio, F. and Molinari, D., A multiscale approach to the simulation of asphaltenes. Computational and Theoretical Chemestry. 975, pp. 76-82, 2011. http://dx.doi.org/10.1016/j.comptc.2011.03.013. [ Links ]
[7] Carauta-A. N.M., Seidl-P. R. and Correia-J. C.G., Computational Simulation of Asphaltene, in Mercosur Congress on Chemical Engineering (2nd, 2005, Brasil) Mercosur Congress on Process Systems Engineering (4th, 2005, Brasil). Enpromer, Rio de Janeiro, Brasil, 2005, pp. 1-6, 2005. [ Links ]
[8] Sedghi, M., Goual, L., Welch, W. and Kubelka, J., Effect of asphaltene structure on association and aggregation using molecular dynamics. The journal of physical chemestry. B. 117 (18), pp. 5765-5776, 2013. http://dx.doi.org/10.1021/jp401584u. [ Links ]
[9] Mullins, O.C., The modified Yen model. Energy & Fuels. 24 (4), pp. 2179-2207, 2010. http://dx.doi.org/10.1021/ef900975e. [ Links ]
[10] Mullins, O.C., Sabbah, H., Pomerantz, A.E., Andrews, A.B., Ruiz-Morales, Y., Mostow, F., Mcfarlane, R., Goual, L., Lepkowicz, R., Cooper, T., Orbulescu, J., Leblanc, R.M., Edwards, J. and Zare, R.N., Advances in asphaltene science and the Yen - Mullins model. Energy & Fuels, 26, pp. 3986-4003, 2012. http:// dx.doi.org/10.1021/ef300185p. [ Links ]
[11] Gray, M.R., Tykwinski, R.R., Stryker, J.M. and Tan, X., Supramolecular assembly model for aggregation of petroleum asphaltenes. Energy & Fuels, 25 (7) , pp. 3125-3134, 2011. http://dx.doi.org/10.1021/ef200654p. [ Links ]
[12] Navarro, L., Álvarez, M., Grosso, J.L. y Navarro, U., Separación y caracterización de resinas y asfaltenos provenientes del crudo castilla. Evaluación de su interacción molecular. CT&F - Ciencia Tecnología y Futuro, 2 (5), pp. 53-67, 2004. [ Links ]
[13] Tharanivasan - A.K., Asphaltene precipitation from crude oil blends , conventional oils, and oils with emulsified water, Dr. Thesis (in Philosophy), University of Calgary, Canada, 211 P.2012, [ Links ]
[14] Plimpton, S., Fast parallel algorithms for short - range molecular dynamics. Journal of Computational Physics, 117, pp. 1-19, 1995. [ Links ]
[15] Rappé, A.K. and Goddard III, W.A., Charge equilibration for molecular dynamics simulations. The journal of physical chemestry, 95 (8), pp. 3358-3363, 1991. [ Links ]
[16] Levitt, M., Hirshberg, M., Sharon, R. and Daggett, V., Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Computer Physics Communications, 91, pp. 215-231, 1995. [ Links ]
[17] Rogel, E., Simulation of interactions in asphaltene aggregates. Energy & Fuels, 14, pp. 566-574, 2000. http://dx.doi.org/10.1021/ef990166p. [ Links ]
J. De León, received the Bsc. in Petroleum Engineering in 2010 and MSc. in Chemical Engineering in 2014, all of them from the Universidad Nacional de Colombia. Currently she is pursuing a doctoral degree in Energy Systems at the same University. Her research interests include molecular simulation, modeling asphaltene aggregation and viscosity and molecular representation of hydrocarbon systems.
B. A. Hoyos, received the Bsc. In 1994 and MSc. In 2003 degrees both in Chemical Engineering and the PhD degree in Energy Systems in 2010, all of them from the Universidad Nacional de Colombia, Medellín, Colombia. Currently he is a Full Professor in the Departamento de Procesos y Energía, Facultad de Minas, Universidad Nacional de Colombia, Medellín, Colombia. His research interests include molecular simulation, modeling asphaltene aggregation and viscosity, modelling wettability alteration by surfactants and specific adsorption.
W.A. Cañas-Marín, received the Bsc. In 1997 from the Universidad de Antioquia, Colombia and MSc. in 2002 degree from the Universidad Industrial de Santander, Colombia, both in Chemical Engineering. He is currently a chemical Engineer at the Instituto Colombiano del Petróleo-ICP of ECOPETROL, S.A. His research interests include thermodynamic characterization of reservoir fluids and phase behavior modeling, effects of external fields on matter, molecular simulation and flow assurance.
